|
Ek I GENEL GÜVENLİLİK VE PERFORMANS GEREKLİLİKLERİ
I. BÖLÜM GENEL GEREKLİLİKLER
1. Cihazlar; imalatçıları tarafından amaçlanan performansı gerçekleştirir ve normal kullanım koşulları altında kullanım amaçlarına uygun olacak şekilde tasarlanır ve imal edilir. Genel olarak kabul görmüş en son teknolojik gelişmeler dikkate alınmak suretiyle; cihazlar güvenli ve etkili olur, cihazların kullanımıyla ilişkili olabilecek risklerin hastaya sağlanan yararlar ile kıyaslandığında kabul edilebilir olması ve sağlık ile güvenliğin yüksek seviyede korunmasına uygun olması sağlanarak, cihazlar hastaların ya da kullanıcıların veya uygun olduğu hallerde diğer kişilerin sağlık ve güvenliğini tehlikeye atmaz. 2. Riskleri mümkün olduğunca azaltmaya yönelik olarak bu Ekte belirtilen gereklilik; fayda-risk oranını olumsuz bir şekilde etkilemeksizin risklerin mümkün olduğu kadar azaltılması anlamına gelir. 3. İmalatçılar, bir risk yönetim sistemi kurar, uygular, dokümante eder ve sürdürür. Risk yönetimi, düzenli sistematik güncelleme gerektiren, cihazın tüm yaşam döngüsü boyunca sürekli tekrarlanan bir süreç olarak anlaşılır. İmalatçılar, risk yönetimini yürütürken: a) Her cihaz için bir risk yönetim planı oluşturur ve dokümante eder, b) Her cihazla ilişkili bilinen ve öngörülebilir tehlikeleri tanımlar ve analiz eder, c) Amaçlanan kullanım esnasında ve makul bir şekilde öngörülebilir hatalı kullanım sırasında ortaya çıkan ve amaçlanan kullanımla ilişkili olan riskleri tahmin eder ve değerlendirir, ç) Bu Ekin 4 numaralı maddesinde belirtilen gereklilikler uyarınca (c) bendinde atıfta bulunulan riskleri ortadan kaldırır veya kontrol eder, d) Üretim aşamasından ve özellikle piyasaya arz sonrası gözetim sisteminden elde edilen bilgilerin; tehlikeler ve bunların meydana gelme sıklığı üzerindeki etkisini, tehlikelerle ilişkili risklerin tahmini üzerindeki etkisini ve toplam risk, fayda-risk oranı ve riskin kabul edilebilirliği üzerindeki etkisini değerlendirir ve e) (d) bendinde atıfta bulunulan bilgilerin etkisiyle ilgili değerlendirmeye dayanarak, gerektiğinde bu Ekin 4 numaralı maddesinde belirtilen gereklilikler doğrultusunda kontrol tedbirlerini tadil eder. 4. Cihazların tasarımına ve imalatına yönelik imalatçılar tarafından kabul edilen risk kontrol tedbirleri, genel kabul görmüş en son teknolojik gelişmeler dikkate alınmak suretiyle güvenlilik ilkelerine uygun olur. Riskleri azaltmak amacıyla, toplam artık riskin yanı sıra her bir tehlikeyle ilişkili artık riskin kabul edilebilir olarak değerlendirilmesi için imalatçılar riskleri yönetir. İmalatçılar; en uygun çözümleri seçerken, aşağıdaki öncelik sırasına uymak suretiyle: a) Güvenli tasarım ve imalat yoluyla riskleri mümkün olduğu kadar ortadan kaldırır veya azaltır, b) Uygun olduğu hallerde, ortadan kaldırılamayan risklere ilişkin olarak gerektiğinde alarmlar dâhil olmak üzere yeterli koruma tedbirleri alır ve c) Güvenliliğe yönelik bilgiler (uyarılar/önlemler/kontrendikasyonlar) ile uygun olduğu hallerde kullanıcılara eğitim sağlar. İmalatçılar, kullanıcıları artık riskler hakkında bilgilendirir. 5. İmalatçılar; kullanım hatasıyla ilgili riskleri ortadan kaldırırken veya azaltırken; a) Cihazın ergonomik özellikleriyle ve cihazın kullanılmasının amaçlandığı ortamla ilgili riskleri mümkün olduğu kadar azaltır (hasta güvenliğine yönelik tasarım) ve b) Hedeflenen kullanıcıların; teknik bilgisini, deneyimini, öğrenimini, eğitimini, uygulanabildiği hallerde kullanım ortamını ve tıbbi ve fiziksel koşullarını göz önünde bulundurur (meslekten olmayan, profesyonel, engelli veya diğer kullanıcılara yönelik tasarım). 6. İmalatçı tarafından belirtildiği şekilde bir cihazın karakteristikleri ve performansı; cihaz normal kullanım koşulları altında meydana gelebilen zorlamalara tabi tutulduğunda ve cihazın imalatçının talimatlarına göre düzenli olarak bakımı yapıldığında, söz konusu cihazın kullanım ömrü süresince hastanın veya kullanıcının ve diğer kişilerin sağlık veya güvenliğini tehlikeye atacak derecede olumsuz olarak etkilenmez. 7. Cihazlar, imalatçı tarafından sağlanan talimatlar ve bilgiler dikkate alınarak nakledilmesi ve depolanması sırasında (örneğin, sıcaklık ve nem dalgalanmaları nedeniyle) öngörülen kullanımları süresince karakteristikleri ve performansı olumsuz olarak etkilenmeyecek şekilde tasarlanır, imal edilir ve ambalajlanır. 8. Bilinen ve öngörülebilir tüm riskler ve istenmeyen yan etkiler; en aza indirgenir ve normal kullanım koşulları altında cihazın gerçekleşen performansından kaynaklanan, hasta ve/veya kullanıcı için ölçülen faydalar ile kıyaslandığında kabul edilebilir olur. 9. Ek XVI’da atıfta bulunulan cihazlar için bu Ekin 1 ve 8 numaralı maddelerinde belirtilen genel güvenlilik gereklilikleri; cihazın belirlenen şartlar altında ve öngörülen amaçlar için kullanıldığında ürün kullanımıyla ilişkili, hiçbir risk teşkil etmemesi ya da kişilerin güvenliğinin ve sağlığının yüksek seviyede korunmasına uygun olarak azami kabul edilebilir riskten fazla olmayan bir risk teşkil etmesi anlamına gelir.
II. BÖLÜM TASARIM VE İMALAT İLE İLGİLİ GEREKLİLİKLER 10. Kimyasal, fiziksel ve biyolojik özellikler 10.1. Cihazlar; bu Ekin I. Bölümünde atıfta bulunulan karakteristiklerin ve performans gerekliliklerinin yerine getirilmesini sağlayacak şekilde tasarlanır ve imal edilir. Aşağıdakilere özellikle dikkat edilir: a) Özellikle toksisiteyle ve ilgili olduğu hallerde alevlenirlikle ilgili olarak kullanılan materyallerin ve maddelerin seçimi, b) Cihazın kullanım amacı ve ilgili olduğu hallerde absorbsiyon, dağılım, metabolizma ve atılım dikkate alınarak, kullanılan materyaller ve maddeler ile biyolojik dokular, hücreler ve vücut sıvıları arasındaki uyumluluk, c) Birden fazla implante edilebilir parçadan oluşan bir cihazın farklı parçaları arasındaki uyumluluk, ç) Süreçlerin materyal özellikleri üzerindeki etkisi, d) Uygun olduğu hallerde, geçerliliği önceden kanıtlanmış biyofiziksel veya modelleme araştırma sonuçları, e) Uygun olduğu hallerde, mukavemet, düktilite, kırılma direnci, aşınma direnci ve yorulma direnci gibi hususlar dikkate alınarak kullanılan materyallerin mekanik özellikleri, f) Yüzey özellikleri ve g) Cihazın, tanımlanmış kimyasal ve/veya fiziksel spesifikasyonları karşıladığının teyidi. 10.2. Cihazlar; kullanım amaçları dikkate alınarak hastalar ve cihazların nakliyesinde, depolanmasında ve kullanılmasında yer alan kişiler için kontaminantlardan ve kalıntılardan kaynaklanan riskleri en aza indirecek şekilde tasarlanır, imal edilir ve ambalajlanır. Bu kontaminantlara ve kalıntılara maruz kalan dokular ile maruz kalma süresine ve sıklığına özellikle dikkat edilir. 10.3. Cihazlar; gazlar dâhil olmak üzere, öngörülen kullanımları süresince temas ettikleri malzemeler ve maddelerle güvenli olarak kullanılabilecekleri bir şekilde tasarlanır ve imal edilir. Tıbbi ürünleri tatbik etme amaçlı cihazlar; bu tıbbi ürünlere ilişkin hükümlere ve kısıtlamalara uygun olarak ilgili tıbbi ürünlerle uyumlu olacak şekilde ve hem tıbbi ürünlerin hem de cihazların performansının, bunların ilgili endikasyonlarına ve kullanım amaçlarına uygun olarak sürdürüleceği bir şekilde tasarlanır ve imal edilir. 10.4. Maddeler 10.4.1. Cihazların tasarımı ve imalatı Cihazlar; aşınma kalıntısı, bozunma ürünleri ve işlem kalıntıları dâhil olmak üzere cihazdan salınabilecek maddelerden veya partiküllerden kaynaklanan riskleri mümkün olduğu kadar azaltacak bir şekilde tasarlanır ve imal edilir. - İnvaziv olan ve insan vücuduna doğrudan temas eden, - Tıbbi ürünleri, vücut sıvılarını veya gazlar dâhil olmak üzere diğer maddeleri tek seferde ya da tekrarlayarak vücuda tatbik eden veya vücuttan uzaklaştıran ya da - Tek seferde ya da tekrarlayarak vücuda tatbik edilecek bu tür tıbbi ürünleri, vücut sıvılarını veya gazlar dâhil olmak üzere maddeleri taşıyan veya depolayan cihazlar veya bunların parçaları ya da bu cihazlarda kullanılan materyaller; yalnızca 10.4.2 uyarınca gerekçelendirildiği takdirde, aşağıdaki maddeleri ağırlıkça yüzdeleri (w/w) % 0,1’in üzerinde bir konsantrasyonda içerir: a) 11/12/2013 tarihli ve 28848 mükerrer sayılı Resmî Gazete’de yayımlanan Maddelerin ve Karışımların Sınıflandırılması, Etiketlenmesi ve Ambalajlanması Hakkında Yönetmeliğin Ek-6’sının 3. Bölümü uyarınca, 1A veya 1B kategorisinin kanserojen, mutajen veya üreme sistemine toksik etki gösteren maddeleri (CMR) veya b) İnsan sağlığı üzerine muhtemel ciddi etkilerine dair bilimsel kanıt bulunan ve 23/6/2017 tarihli ve 30105 sayılı Resmî Gazete’de yayımlanan Kimyasalların Kaydı, Değerlendirilmesi, İzni ve Kısıtlanması Hakkında Yönetmeliğin 49 uncu maddesinde belirtilen prosedüre uygun olarak ya da Komisyon tarafından bir tasarruf kabul edildiğinde söz konusu tasarrufta belirlenen kriterler arasında insan sağlığıyla ilgili olan kriterlere uygun olarak tanımlanan endokrin bozucu özelliklere sahip maddeleri. 10.4.2. CMR ve/veya endokrin bozucu maddelerin mevcut olmasıyla ilgili gerekçelendirme Bu tür maddelerin mevcudiyetine yönelik gerekçelendirme, aşağıdaki hususlara dayanır: a) Maddeye maruz kalması muhtemel hasta ve kullanıcıların analizi ve tahmini, b) Mevcut olduğu durumda; bağımsız araştırmalar, hakem denetimli çalışmalar, ilgili bilimsel komitelerden alınan bilimsel görüşler hakkında bilgiler de dâhil olmak üzere, olası alternatif maddelerin, materyallerin veya tasarımların analizi ve bu tür alternatiflerin mevcut olmasıyla ilgili analiz, c) Cihazların amaçlanan kullanımının, çocukların ya da gebe veya emziren kadınların tedavisini ya da bu tür maddeler ve/veya materyaller açısından özellikle etkilenebilir olduğu düşünülen diğer hasta gruplarının tedavisini içerip içermediğinin dikkate alınması dâhil olmak üzere, mevcutsa olası madde ve/veya materyal ikamelerinin veya yapılabilir ise tasarım değişikliklerinin; ürünün işlevselliğinin, performansının ve fayda-risk oranlarının sürdürülmesi bakımından neden uygun olmadığına yönelik tartışma ve ç) Uygulanabildiği hallerde ve mevcut olduğu durumda, bu Ekin 10.4.3 ve 10.4.4 numaralı maddeleri uyarınca AB Komisyonunun ilgili bilimsel komitesince hazırlanan güncel rehberleri. 10.4.3. Ftalatlara ilişkin kılavuzlar Bu Ekin 10.4.1 numaralı maddesinin (a) ve (b) bentlerinde atıfta bulunulan madde gruplarından herhangi birinde ftalatların mevcut olmasıyla ilgili bir fayda-risk değerlendirmesini kapsayan AB Komisyonu’nun ilgili bilimsel komitesince hazırlanan rehberler ve güncellemeleri dikkate alınır. 10.4.4. Diğer CMR ve endokrin bozucu maddelere ilişkin kılavuzlar Uygun olduğu hallerde, bu Ekin 10.4.1 numaralı maddesinin (a) ve (b) bentlerinde atıfta bulunulan diğer maddeler için de 10.4.3 numaralı maddede atıfta bulunulduğu şekilde AB Komisyonunun ilgili bilimsel komitesince hazırlanan rehberler dikkate alınır 10.4.5. Etiketleme Bu Ekin 10.4.1 numaralı maddesinde atıfta bulunulduğu şekilde, cihazların, bu cihazların parçalarının veya bu cihazlarda kullanılan materyallerin, 10.4.1 numaralı maddesinin (a) ve (b) bentlerinde atıfta bulunulan maddeleri ağırlıkça yüzdeleri (w/w) %0,1’in üzerinde bir konsantrasyonda içermesi durumunda, bu tür maddelerin mevcut olduğunu gösteren ve söz konusu maddelerin listesini içeren etiketler, cihazın kendisine ve/veya her bir birim ambalajına ya da uygulanabildiği hallerde satış ambalajına iliştirilir. Bu tür cihazların amaçlanan kullanımı, çocukların tedavisini ya da gebe veya emziren kadınların tedavisini ya da bu tür maddeler ve/veya materyaller açısından özellikle etkilenebilir olduğu düşünülen diğer hasta gruplarının tedavisini içeriyorsa, bu hasta gruplarına yönelik artık risklere ve mevcut ise uygun ihtiyati tedbirlere ilişkin bilgiler, kullanım talimatında verilir. 10.5. Cihazlar, cihaz ve kullanılmasının amaçlandığı ortamın niteliği dikkate alınarak cihaz içerisine istenmeyen bir şekilde maddelerin girmesinden kaynaklanan riskleri mümkün olduğu kadar azaltacak şekilde tasarlanır ve imal edilir. 10.6. Yalnızca sağlam deriye temas eden cihazlar hariç olmak üzere cihazlar, hastanın veya kullanıcının vücuduna salınan ya da salınabilecek olan partiküllerin boyutu ve özellikleriyle bağlantılı riskleri mümkün olduğu kadar azaltacak şekilde tasarlanır ve imal edilir. Nanomateryallere özellikle dikkat edilir. 11. Enfeksiyon ve mikrobiyal kontaminasyon 11.1. Cihazlar ve bunların imalat süreçleri; hastalara, kullanıcılara ve gerektiğinde diğer kişilere yönelik enfeksiyon riskini ortadan kaldıracak veya mümkün olduğu kadar azaltacak şekilde tasarlanır. Tasarım: a) İstenmeyen kesiklerden ve batmalardan kaynaklanan, örneğin iğne batması yaralanmaları gibi, riskleri mümkün olduğu kadar ve uygun bir şekilde azaltır, b) Kolay ve güvenli kullanımı mümkün kılar, c) Cihazdan kaynaklanan mikrobiyal sızıntıları ve/veya kullanım süresince mikrobiyal maruz kalmayı mümkün olduğu kadar azaltır ve ç) Cihazın ya da örnekler veya sıvılar gibi cihaz içeriğinin mikrobiyal kontaminasyonunu önler. 11.2. Gerektiği takdirde cihazlar; güvenli bir şekilde temizliklerini, dezenfeksiyonlarını ve/veya yeniden sterilizasyonlarını kolaylaştıracak biçimde tasarlanır. 11.3. Spesifik bir mikrobiyal duruma sahip olarak etiketlenen cihazlar, piyasaya arz edildiklerinde ve imalatçı tarafından belirlenen nakliye ve depolama şartları altında, bu durumlarını koruyacak şekilde tasarlanır, imal edilir ve ambalajlanır. 11.4. Steril durumda tedarik edilen cihazlar, piyasaya arz edildiklerinde steril olmalarını sağlamak amacıyla, steril durumlarını korumaya yönelik ambalaj zarar görmediği sürece, imalatçı tarafından belirlenen nakliye ve depolama şartları altında ambalaj, kullanım noktasında açılıncaya kadar steril kalmalarını sağlamak üzere uygun prosedürler uyarınca tasarlanır, imal edilir ve ambalajlanır. Bu ambalajın bütünlüğünün son kullanıcı için açıkça görülebilir olması sağlanır. 11.5. Steril olarak etiketlenen cihazlar, geçerli kılınmış uygun yöntemler vasıtasıyla işlenir, imal edilir, ambalajlanır ve sterilize edilir. 11.6. Steril edilmesi amaçlanan cihazlar, uygun ve kontrollü şartlarda ve tesislerde imal edilir ve ambalajlanır. 11.7. Steril olmayan cihazlara yönelik ambalajlama sistemleri; ürünün bütünlüğünü ve temizliğini korur ve cihazların kullanımdan önce steril edilecek olması halinde mikrobiyal kontaminasyon riskini en aza indirir. Bu ambalajlama sistemi, imalatçı tarafından belirtilen sterilizasyon metodu için uygun olur. 11.8. Cihazın etiketlemesi; cihazların steril olduğunu göstermek için kullanılan sembole ilave olarak hem steril hem de steril olmayan durumda piyasaya arz edilen aynı veya benzer cihazları birbirinden ayırır. 12. Tıbbi ürün olduğu kabul edilen bir madde ihtiva eden cihazlar ve insan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar 12.1. 1 inci maddenin altıncı fıkrasının ikinci cümlesinde atıfta bulunulan cihazlar söz konusu olduğunda, ayrı olarak kullanıldığında Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin 4 üncü maddesinin (b) bendi çerçevesinde bir tıbbi ürün olduğu kabul edilen maddenin kalitesi, güvenliliği ve yararlılığı; bu Yönetmelik kapsamında uygulanabilir uygunluk değerlendirme prosedürünün gerektirdiği şekilde, aynı Yönetmeliğin Ek 1’inde belirtilen metotlarla analoji yapılarak doğrulanır. 12.2. İnsan vücuduna girmesi amaçlanan ve insan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar; bu Yönetmelik kapsamında uygulanabilir uygunluk değerlendirme işleminin gerektirdiği şekilde, absorbsiyonun, dağılımın, metabolizmanın, atılımın, lokal toleransın, toksisitenin; diğer cihazlarla, tıbbi ürünlerle veya diğer maddelerle etkileşimin ve advers reaksiyonlar potansiyelinin değerlendirilmesi için bu Yönetmeliğin kapsamadığı konularla sınırlı olmak üzere uygulanabildiği hallerde Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin Ek 1’inde belirtilen ilgili gerekliliklere uyar. 13. Biyolojik kaynaklı materyaller ihtiva eden cihazlar 13.1. 1 inci maddenin üçüncü fıkrasının (ç) bendi uyarınca bu Yönetmeliğin kapsadığı, cansız veya cansız hale getirilmiş insan kaynaklı dokuların veya hücrelerin türevleri kullanılarak imal edilen cihazlar için aşağıdakiler uygulanır: a) Dokuların ve hücrelerin; bağışlanması, temin edilmesi ve test edilmesi İnsan Doku ve Hücreleri ile Bunlarla İlgili Merkezlerin Kalite ve Güvenliği Hakkında Yönetmelik uyarınca yapılır, b) Bu dokuların ve hücrelerin veya bunların türevlerinin işlenmesi, muhafaza edilmesi ve ilgili diğer işlemler; hastaların, kullanıcıların ve gerektiğinde diğer kişilerin güvenliğini sağlayacak şekilde yürütülür. Özellikle virüsler ve diğer bulaşıcı ajanlar açısından güvenlilik; uygun kaynak bulma yöntemleriyle ve imalat süreci boyunca geçerli kılınmış eliminasyon veya inaktivasyon yöntemlerinin uygulanmasıyla ele alınır, c) Bu cihazlara yönelik izlenebilirlik sistemi; İnsan Doku ve Hücreleri ile Bunlarla İlgili Merkezlerin Kalite ve Güvenliği Hakkında Yönetmelik ve 11/4/2007 tarihli ve 5624 sayılı Kan ve Kan Ürünleri Kanunu ve 4/12/2008 tarihli ve 27074 sayılı Kan Ürünleri Yönetmeliğinde belirtilen izlenebilirlik ve veri koruma gereklilikleri ile uyumlu olur ve bu gereklilikleri tamamlayıcıdır. 13.2. Cansız veya cansız hale getirilmiş hayvan kaynaklı dokular veya hücreler ya da bunların türevleri kullanılarak imal edilen cihazlar için aşağıdakiler uygulanır: a) Uygulanabilir olduğunda hayvan türleri dikkate alınarak, hayvan kaynaklı dokular ve hücreler ya da bunların türevleri, dokuların amaçlanan kullanımına uygun olan veteriner kontrollerine tabi tutulmuş hayvanlardan elde edilir. Hayvanların coğrafi kaynağına ilişkin bilgiler imalatçı tarafından muhafaza edilir, b) Hayvan kaynaklı dokuların, hücrelerin ve maddelerin veya bunların türevlerinin tedarik edilmesi, işlenmesi, muhafaza edilmesi, test edilmesi ve kullanımı; hastaların, kullanıcıların ve gerektiğinde diğer kişilerin güvenliğini sağlayacak şekilde yürütülür. Özellikle virüsler ve diğer bulaşıcı ajanlar açısından güvenlilik, imalat süreci boyunca geçerli kılınmış eliminasyon veya viral inaktivasyon yöntemlerinin uygulanmasıyla ele alınır; ancak bu husus, söz konusu yöntemlerin kullanımının cihazın klinik yararına gölge düşüren kabul edilemez bozulmaya yol açabileceği durumlarda geçerli değildir. c) 14/1/2021 tarihli ve 31364 sayılı Resmî Gazete’de yayımlanan Hayvan Kaynaklı Dokular Kullanılarak İmal Edilen Tıbbi Cihazlara Dair Yönetmeliğin atıfta bulunulduğu şekilde, hayvan kaynaklı dokular ve hücreler ya da bunların türevleri kullanılarak imal edilen cihazlar söz konusu olduğunda, aynı Yönetmeliğin belirtilen özel gereklilikleri uygulanır. 13.3. Bu Ekin 13.1 ve 13.2 numaralı maddelerinde atıfta bulunulanlar haricindeki cansız biyolojik maddeler kullanılarak imal edilen cihazlar için, atıkların bertarafı dâhil olmak üzere bu maddelerin işlenmesi, muhafaza edilmesi, test edilmesi ve kullanımı; hastaların, kullanıcıların ve gerektiğinde diğer kişilerin güvenliğini sağlayacak şekilde yürütülür. Özellikle virüsler ve diğer bulaşıcı ajanlar açısından güvenlilik; uygun kaynak bulma yöntemleriyle ve imalat süreci boyunca geçerli kılınmış eliminasyon veya inaktivasyon yöntemlerinin uygulanmasıyla ele alınır. 14. Cihazların yapısı ve çevre ile etkileşimi 14.1. Cihazın, başka bir cihazla veya ekipmanla birlikte kullanımı amaçlanmışsa bağlantı sistemi dâhil olmak üzere tüm kombinasyon güvenli olur ve cihazların belirtilen performansını etkilemez. Bu tür kombinasyonlara uygulanan kullanım kısıtlamaları, etiket ve/veya kullanım talimatında belirtilir. Sıvı veya gaz transferi, elektriksel veya mekanik bağlantı gibi kullanıcının yapması gereken bağlantılar, hatalı bağlantı gibi olası tüm riskleri en aza indirecek şekilde tasarlanır ve yapılır. 14.2. Cihazlar, aşağıdaki riskleri ortadan kaldıracak veya mümkün olduğu kadar azaltacak şekilde tasarlanır ve imal edilir: a) Hacim/basınç oranı, boyutsal ve uygun olduğu hallerde ergonomik özellikler dâhil olmak üzere cihazların fiziksel özellikleriyle bağlantılı yaralanma riski, b) Manyetik alanlar, harici elektriksel ve elektromanyetik etkiler, elektrostatik boşalma, diyagnostik veya terapötik prosedürler ile ilişkili radyasyon, basınç, nem, sıcaklık, basınç değişiklikleri ve ivme ya da radyo sinyali girişimleri (interferans) gibi makul bir şekilde öngörülebilir dış etkiler veya çevresel koşullarla bağlantılı riskler, c) Gazlar dâhil olmak üzere materyallerle, sıvılarla ve maddelerle temas ettiğinde, cihazın kullanımıyla ilişkili ve normal kullanım koşulları altında maruz kalınan riskler, ç) Yazılım ile içinde çalıştığı ve etkileşimde bulunduğu bilgi teknolojileri (BT) ortamı arasındaki olası olumsuz etkileşimle ilişkili riskler, d) Cihaz içerisine kazara maddelerin girmesi ile ilgili riskler, e) Tanı veya uygulanan tedavide genel olarak kullanılan diğer cihazlarla karşılıklı etkileşim riskleri, f) Bakım veya kalibrasyonun mümkün olmadığı durumda (implantlarda olduğu gibi), kullanılan materyallerin yaşlanmasından ya da herhangi bir ölçüm veya kontrol mekanizmasının doğruluğunu yitirmesinden kaynaklanan riskler. 14.3. Cihazlar; normal kullanım sırasında ve tek hata durumunda, yanma veya patlama riskini en aza indirecek şekilde tasarlanır ve imal edilir. Cihazların amaçlanan kullanımı; alevlenebilir veya patlayıcı maddelere ya da yanmaya neden olabilecek maddelere maruz kalmayı veya söz konusu maddelerle birlikte kullanımı içeriyorsa, bu cihazlara özellikle dikkat edilir. 14.4. Cihazlar; ayarlamanın, kalibrasyonun ve bakımın güvenli ve etkili biçimde yapılabileceği bir şekilde tasarlanır ve imal edilir. 14.5. Başka cihazlarla veya ürünlerle birlikte çalışması amaçlanan cihazlar; birlikte çalışabilirlikleri ve uyumlulukları güvenilir ve güvenli olacak şekilde tasarlanır ve imal edilir. 14.6. Herhangi bir ölçme, izleme veya gösterge skalası; kullanım amacı, kullanıcılar ve cihazların kullanılmasının amaçlandığı çevre koşulları dikkate alınarak ergonomik prensipler doğrultusunda tasarlanır ve imal edilir. 14.7. Cihazlar; kullanıcı, hasta veya diğer kişiler tarafından cihazların ve ilgili atık maddelerin güvenli olarak bertarafını kolaylaştıracak şekilde tasarlanır ve imal edilir. İmalatçılar bu amaçla, cihazlarının kullanım sonrasında güvenli bir şekilde bertarafına yönelik prosedürleri ve tedbirleri tanımlar ve test eder. Bu tür prosedürler kullanım talimatında açıklanır. 15. Diyagnostik veya ölçüm fonksiyonu olan cihazlar 15.1. Diyagnostik cihazlar ve ölçüm fonksiyonu olan cihazlar; uygun bilimsel ve teknik yöntemlere dayanarak kullanım amaçları bakımından; yeterli doğruluk, kesinlik ve stabilite sağlanacak şekilde tasarlanır ve imal edilir. Doğruluk limitleri imalatçı tarafından belirtilir. 15.2. Ölçüm fonksiyonu olan cihazlarla yapılan ölçümler, 21/6/2002 tarihli ve 24792 sayılı Resmî Gazete’de yayımlanan Uluslararası Birimler Sistemine Dair Yönetmeliğe uygun olan birimlerle ifade edilir. 16. Radyasyondan koruma 16.1. Genel a) Cihazlar; diyagnostik ve terapötik amaçlar için tanımlanmış uygun dozların uygulanmasını kısıtlamaksızın, kullanım amacıyla uyumlu olarak hastaların, kullanıcıların ve diğer kişilerin radyasyona maruz kalmasını mümkün olduğunca azaltacak şekilde tasarlanır, imal edilir ve ambalajlanır. b) Tehlikeli veya potansiyel olarak tehlikeli radyasyon yayan cihazlara yönelik kullanım talimatları; yayılan radyasyonun niteliği, hastayı ve kullanıcıyı koruma yolları, hatalı kullanımı önleme yolları ve kurulumdan kaynaklanan riskleri mümkün ve uygun olduğu kadarıyla azaltma yolları hakkında ayrıntılı bilgiler içerir. Kabul ve performans testleri, kabul kriterleri ve bakım prosedürü hakkındaki bilgiler de belirtilir. 16.2. Planlanan radyasyon a) Faydasının emisyondan kaynaklanan risklerden daha fazla olduğu kabul edilen ve spesifik bir tıbbi amaç için tehlikeli ya da potansiyel olarak tehlikeli dozlarda iyonlaştırıcı ve/veya iyonlaştırıcı olmayan radyasyon yaymak üzere tasarlanan cihazlar söz konusu olduğunda, kullanıcının emisyonları kontrol etmesi mümkün olur. Bu tür cihazlar, kabul edilebilir bir tolerans dâhilinde ilgili değişken parametrelerin yeniden üretilebilirliğini sağlayacak şekilde tasarlanır ve imal edilir. b) Tehlikeli veya potansiyel olarak tehlikeli iyonlaştırıcı ve/veya iyonlaştırıcı olmayan radyasyon yayması amaçlanan cihazlar söz konusu olduğunda, bu cihazlar mümkün olduğu durumlarda bu tür emisyonlarla ilgili görsel ve/veya sesli uyarı sistemleriyle donatılır. 16.3. Cihazlar; hastaların, kullanıcıların ve diğer kişilerin istenmeyen, sızıntı veya saçılan radyasyona maruz kalmasını mümkün olduğu kadar azaltacak şekilde tasarlanır ve imal edilir. Mümkün ve uygun olduğu hallerde; hastaların, kullanıcıların ve etkilenebilecek diğer kişilerin radyasyona maruz kalmasını azaltan yöntemler seçilir. 16.4. İyonlaştırıcı radyasyon a) İyonlaştırıcı radyasyon yayması amaçlanan cihazlar; iyonlaştırıcı radyasyona maruz kalmadan kaynaklı tehlikelere karşı korunmaya yönelik temel güvenlik standartları dikkate alınarak tasarlanır ve imal edilir. b) İyonlaştırıcı radyasyon yayması amaçlanan cihazlar; mümkün olduğu hallerde, kullanım amacı dikkate alınmak suretiyle yayılan radyasyonun niceliğinin, geometrisinin ve niteliğinin değiştirilebilmesini ve kontrol edilebilmesini ve mümkünse tedavi boyunca izlenebilmesini sağlayacak şekilde tasarlanır ve imal edilir. c) Tanısal radyoloji amaçlı iyonlaştırıcı radyasyon yayan cihazlar; hastanın ve kullanıcının radyasyona maruz kalmasını en aza indirirken, hedeflenen tıbbi amaca uygun bir görüntü ve/veya çıktı kalitesi sağlayacak şekilde tasarlanır ve imal edilir. ç) İyonlaştırıcı radyasyon yayan ve terapötik radyoloji amaçlı cihazlar; verilen dozun, ışın tipinin, enerjinin ve uygun olduğu hallerde radyasyon kalitesinin güvenilir bir biçimde izlenmesini ve kontrol edilmesini sağlayacak şekilde tasarlanır ve imal edilir. 17. Elektronik programlanabilir sistemler – elektronik programlanabilir sistemler içeren cihazlar ve kendi başına cihaz olan yazılımlar 17.1. Yazılımlar dâhil olmak üzere elektronik programlanabilir sistemler içeren cihazlar ya da kendi başına cihaz olan yazılımlar; tekrarlanabilirliği, güvenilirliği sağlamak ve amaçlanan kullanımları doğrultusunda performans göstermek üzere tasarlanır. Tek hata durumu halinde, ortaya çıkan riskleri veya performans bozukluğunu gidermek ya da mümkün olduğu kadar azaltmak için uygun önlemler alınır. 17.2. Yazılım içeren cihazlar veya kendi başına cihaz olan yazılımlar söz konusu olduğunda, bu yazılımlar; geliştirme yaşam döngüsü, bilgi güvenliği, risk yönetimi, doğrulama ve validasyon ilkeleri dikkate alınmak suretiyle en son teknolojik gelişmelere uygun olarak geliştirilir ve imal edilir. 17.3. Bu Ekin 17 numaralı maddesinde atıfta bulunulan ve mobil bilgi işleme platformlarıyla kombinasyon halinde kullanılması amaçlanan yazılımlar; mobil platformun spesifik özellikleri (örneğin ekranın boyutu ve kontrast oranı) ve kullanımlarıyla ilişkili dış faktörler (ışık veya gürültü seviyesine göre değişen ortam) dikkate alınarak tasarlanır ve imal edilir. 17.4. İmalatçılar; yazılımın amaçlandığı şekilde çalışması için gerekli olan donanım, bilgi teknolojileri (BT) ağ karakteristikleri ve yetkisiz erişime karşı koruma dâhil bilgi teknolojileri (BT) güvenlik önlemleri ile ilgili asgari gereklilikleri belirler. 18. Aktif cihazlar ve onlara bağlanan cihazlar 18.1. İmplante edilebilir olmayan aktif cihazlar için tek hata durumu halinde ortaya çıkan riskleri gidermek veya mümkün olduğu kadar azaltmak için uygun önlemler alınır. 18.2. Hasta güvenliğinin dâhili bir güç kaynağına bağlı olduğu durumlarda cihazlar, güç kaynağının durumunu belirten araçlarla ve güç kaynağı kapasitesinin kritik düzeyde olduğu zamana yönelik uygun bir uyarıyla veya göstergeyle donatılır. Gerektiğinde bu tür uyarı veya gösterge, güç kaynağı kapasitesi kritik düzeye gelmeden önce aktif olur. 18.3. Hasta güvenliğinin harici bir güç kaynağına bağlı olduğu durumlarda cihazlar, herhangi bir güç kesintisini bildirmek için bir alarm sistemi içerir. 18.4. Hastanın bir veya daha fazla klinik parametresini izleme amaçlı cihazlar; ölüme ya da hastanın sağlık durumunda ciddi bozulmaya yol açabilecek durumlarda kullanıcıyı uyarmak üzere uygun alarm sistemleriyle donatılır. 18.5. Cihazlar; söz konusu cihazın veya amaçlanan kullanım ortamındaki diğer cihazların ya da ekipmanın çalışmasını bozabilecek elektromanyetik interferans oluşturma risklerini mümkün olduğu kadar azaltacak şekilde tasarlanır ve imal edilir. 18.6. Cihazlar; amaçlandıkları şekilde çalışmalarını mümkün kılmak için yeterli olan, elektromanyetik interferansa karşı yapısal bir dayanıklılık düzeyi sağlayacak şekilde tasarlanır ve imal edilir. 18.7. Cihazlar; kurulumunun ve bakımının imalatçı tarafından belirtilen şekilde yapılması şartıyla hem cihazın normal kullanımı sırasında hem de cihazdaki tek hata durumu halinde hasta, kullanıcı veya diğer kişiler için kazara meydana gelebilecek elektrik şoku riskini mümkün olduğu kadar önleyecek şekilde tasarlanır ve imal edilir. 18.8. Cihazlar; amaçlandığı şekilde çalışmasını engelleyebilecek yetkisiz erişime karşı, mümkün olduğu kadar korunaklı şekilde tasarlanır ve imal edilir. 19. İmplante edilebilir aktif cihazlar için özel gereklilikler 19.1. İmplante edilebilir aktif cihazlar, aşağıdaki riskleri ortadan kaldıracak veya mümkün olduğu kadarıyla en aza indirecek şekilde tasarlanır ve imal edilir: a) Elektrik kullanılması durumunda özellikle cihazların yalıtımına, kaçak akımlarına ve aşırı ısınmasına yönelik olmak üzere enerji kaynaklarının kullanımıyla bağlantılı riskler, b) Defibrilatörlerin veya yüksek frekanslı cerrahi ekipmanın kullanımından kaynaklanan riskler dâhil olmak üzere tıbbi tedaviyle bağlantılı riskler ve c) Aşağıda belirtilen riskler dâhil olmak üzere bakım ve kalibrasyonun mümkün olmadığı durumlarda ortaya çıkabilecek riskler: - Kaçak akımların aşırı artışı, - Kullanılan materyallerin yaşlanması, - Cihazın aşırı ısı oluşturması, - Ölçüm veya kontrol mekanizmalarının doğruluğunun azalması. 19.2. İmplante edilebilir aktif cihazlar: - Uygulanabildiği hallerde, cihazlar ile bu cihazlarla tatbik edilmesi amaçlanan maddelerin uyumluluğunu ve - Enerji kaynağının güvenilirliğini sağlayacak şekilde tasarlanır ve imal edilir. 19.3. İmplante edilebilir aktif cihazlar ve uygun olduğu hallerde bunların bileşenleri; cihazlarla veya bileşenleriyle bağlantılı potansiyel bir riskin ortaya çıkarılmasını takiben alınacak gerekli önlemleri mümkün kılmak amacıyla tanımlanabilir olur. 19.4. İmplante edilebilir aktif cihazlar, kendilerinin ve imalatçılarının kesin surette tanımlanabileceği (özellikle cihazın tipiyle ve imalat yılıyla ilgili olarak) bir kod taşır. Gerektiği takdirde, cerrahi bir operasyona ihtiyaç duyulmadan bu kodu okumak mümkün olur. 20. Mekanik ve termal risklere karşı koruma 20.1. Cihazlar; harekete karşı direnç, dengesizlik ve hareketli parçalar gibi mekanik risklere karşı hastaları ve kullanıcıları koruyacak şekilde tasarlanır ve imal edilir. 20.2. Cihazlar; belirlenmiş performansın bir parçası olmadığı sürece kaynaktaki titreşimler başta olmak üzere titreşimleri sınırlamaya yönelik uygun araçlar ve teknik ilerlemeler dikkate alınarak, cihazın oluşturduğu titreşimden kaynaklanan riskleri mümkün olan en düşük seviyeye indirecek şekilde tasarlanır ve imal edilir. 20.3. Cihazlar; yayılan gürültü belirlenmiş performansın bir parçası olmadığı sürece, kaynaktaki gürültü başta olmak üzere gürültüyü azaltmaya yönelik uygun araçlar ve teknik ilerlemeler dikkate alınarak, yayılan gürültüden kaynaklanan riskleri mümkün olan en düşük seviyeye indirecek şekilde tasarlanır ve imal edilir. 20.4. Kullanıcının veya diğer kişilerin kullanmak zorunda olduğu elektrik, gaz ya da hidrolik ve pnömatik enerji kaynaklarına yönelik terminaller ve konnektörler, olası tüm riskleri en aza indirecek şekilde tasarlanır ve yapılır. 20.5. Belirli parçaların montajı veya yeniden montajı sırasında yapılması muhtemel olan ve bir risk kaynağı olabilecek hatalar; bu tür parçaların tasarımı ve yapımı yoluyla veya bunun yapılamadığı durumda bu parçaların ve/veya bunların yuvaları üzerinde verilen bilgiler yoluyla engellenir. Aynı bilgiler; bir riski önlemek için hareket yönünün bilinmesinin gerektiği durumlarda, hareketli parçaların ve/veya bunların yuvalarının üzerinde belirtilir 20.6. Isı vermesi veya belirlenen sıcaklıklara ulaşması amaçlanan parçalar ya da alanlar hariç olmak üzere cihazların ulaşılabilir parçaları ve bunların çevresi, normal kullanım koşulları altında potansiyel olarak tehlikeli sıcaklıklara erişmez. 21. Enerji veya madde veren cihazların hasta ya da kullanıcı için oluşturduğu risklere karşı koruma 21.1. Hastaya enerji veya madde veren cihazlar; hastanın veya kullanıcının güvenliğini sağlamak amacıyla, verilecek miktarı yeterince doğru olarak ayarlayabilecek ve sürdürebilecek şekilde tasarlanır ve yapılır. 21.2. Cihazlar; verilen enerji veya madde miktarında bir tehlike oluşturabilecek olan her türlü eksikliği önleme ve/veya gösterme araçlarıyla donatılır. Cihazlar; bir enerji ve/veya madde kaynağından tehlikeli seviyelerde enerji ya da maddenin kazara salınmasını mümkün olduğunca önlemek için uygun araçlar içerir. 21.3. Kontrollerin ve göstergelerin işlevi, cihazların üzerinde açıkça belirtilir. Bir cihazın çalışması için gerekli olan talimatları taşıması ya da görsel bir sistem aracılığıyla çalışma veya ayar parametrelerini göstermesi durumunda, bu tür bilgiler kullanıcı ve uygun olduğunda hasta için anlaşılabilir olur. 22. Meslekten olmayan kişilerin kullanımına yönelik tıbbi cihazların oluşturduğu risklere karşı koruma 22.1. Meslekten olmayan kişilerin kullanımına yönelik cihazlar; bu kişilerin becerileri ve mevcut imkânları ile birlikte söz konusu kişilerin tekniklerinde ve çevrelerinde makul olarak ön görülebilen farklılıklardan kaynaklanan etki dikkate alınarak, kullanım amaçlarına uygun olarak çalışacak şekilde tasarlanır ve imal edilir. İmalatçı tarafından sağlanan bilgilerin ve talimatların, meslekten olmayan kişilerce anlaşılması ve uygulanması kolay olur. 22.2. Meslekten olmayan kişilerin kullanımına yönelik cihazlar: - Gerektiği takdirde uygun eğitim ve/veya bilgilendirme sonrasında, cihazın hedeflenen kullanıcı tarafından prosedürün her aşamasında güvenli ve doğru bir şekilde kullanılabilmesini sağlayacak, - İstenmeyen kesiklerden ve batmalardan kaynaklanan, örneğin iğne batması yaralanmaları gibi, riskleri mümkün ve uygun olduğu kadarıyla azaltacak ve - Cihazın kullanımında ve uygulanabilir olduğu hallerde sonuçların yorumlanmasında hedeflenen kullanıcının hata yapma riskini mümkün olduğu kadar azaltacak şekilde tasarlanır ve imal edilir 22.3. Meslekten olmayan kişilerin kullanımına yönelik cihazlar; uygun olduğu hallerde meslekten olmayan kişinin: - Cihazın imalatçısı tarafından amaçlandığı şekilde çalışacağını kullanım sırasında doğrulayabilmesini ve - Mümkünse, cihaz geçerli bir sonuç vermediği durumda uyarılmasını, sağlayacak bir prosedür içerir.
III. BÖLÜM CİHAZLA BİRLİKTE TEMİN EDİLEN BİLGİLERE İLİŞKİN GEREKLİLİKLER 23. Etiket ve kullanım talimatı 23.1. İmalatçı tarafından temin edilen bilgilere ilişkin genel gereklilikler Her cihazın beraberinde, cihazın ve imalatçısının tanımlanması için gerekli bilgiler ile kullanıcı ya da uygun olduğu takdirde diğer bir kişi için yararlı olabilecek her türlü güvenlilik ve performans bilgileri bulunur. Bu tür bilgiler; aşağıdakiler dikkate alınarak cihazın kendisinin üzerinde, ambalajının üzerinde ya da kullanım talimatında bulunabilir ve imalatçının bir web sitesi varsa bu web sitesinde sunulur ve güncel tutulur: a) Etiket ve kullanım talimatının ortamı, formatı, içeriği, okunabilirliği ve konumu; ilgili cihaza, kullanım amacına ve hedeflenen kullanıcının/kullanıcıların teknik bilgisine, deneyimine, öğrenimine veya eğitimine uygun olur. Özellikle, kullanım talimatı hedeflenen kullanıcı tarafından kolayca anlaşılacak ve uygun olduğunda çizimlerle ve diyagramlarla desteklenecek şekilde yazılır. b) Etikette gerekli bilgiler, cihazın kendisinin üzerinde verilir. Eğer bu uygulanabilir veya uygun değilse, bilgilerin bazıları ya da tamamı, her bir birim ambalajının üzerinde ve/veya çoklu cihaz ambalajının üzerinde bulunabilir. c) Etiketler; insan tarafından okunabilir bir formatta sunulur ve radyo frekans tanımlama (RFID) veya barkodlar gibi makine tarafından okunabilir bilgiler ile desteklenebilir. ç) Kullanım talimatı, cihazlarla birlikte sunulur. İstisna olarak, sınıf I ve sınıf IIa cihazların kullanım talimatları olmadan güvenli bir şekilde kullanılabilmesi mümkün ise ve bu Bölümde aksi belirtilmediği sürece, bu tür cihazlar için kullanım talimatı gerekmez. d) Çoklu cihazların tek bir kullanıcıya ve/veya yere temin edilmesi durumunda, kabul etmesi halinde alıcıya kullanım talimatının tek bir sureti verilebilir; ancak alıcı, bedelsiz sağlanacak daha fazla sureti her koşulda talep edebilir. e) Kullanım talimatı; sadece 2/4/2015 tarihli ve 29314 sayılı Resmî Gazete’de yayımlanan Tıbbi Cihazların Elektronik Kullanım Talimatları Hakkında Tebliğde veya bu Tebliğ kapsamında düzenlenen uygulama kurallarında belirtilen şartlar altında kullanıcıya, kâğıt dışındaki başka bir formatta örneğin elektronik ortamda sunulabilir. f) Kullanıcıya ve/veya diğer kişiye bildirilmesi gereken artık riskler; sınırlamalar, kontrendikasyonlar, önlemler veya uyarılar şeklinde imalatçı tarafından temin edilen bilgilere dâhil edilir. g) Uygun olduğu hallerde imalatçı tarafından temin edilen bilgiler, uluslararası kabul görmüş semboller biçiminde olur. Kullanılan her sembol veya tanımlama rengi, uyumlaştırılmış standartlara ya da ortak spesifikasyonlara uygun olur. Uyumlaştırılmış standartların veya ortak spesifikasyonların bulunmadığı alanlarda, bu semboller ve renkler, cihazla birlikte temin edilen dokümantasyonda açıklanır. 23.2. Etiket üzerindeki bilgiler Etiket aşağıdaki ayrıntıların tümünü taşır: a) Cihazın adı veya ticari adı, b) Bir kullanıcının cihazı tanıması için kesinlikle gerekli olan detaylar, ambalajın içeriği ve kullanıcı için net olmaması halinde cihazın kullanım amacı, c) İmalatçının adı, kayıtlı ticari unvanı veya kayıtlı ticari markası ve kayıtlı işyeri adresi, ç) Uygulanabildiği hallerde, yetkili temsilcinin adı ve kayıtlı işyeri adresi, d) Uygulanabildiği hallerde, cihazın aşağıdakileri içerdiğine veya ihtiva ettiğine dair bir gösterge: - İnsan kanı veya plazması türevi dâhil olmak üzere bir tıbbi madde veya - İnsan kaynaklı dokular veya hücreler ya da bunların türevleri veya - Hayvan Kaynaklı Dokular Kullanılarak İmal Edilen Tıbbi Cihazlara Dair Yönetmelikte belirtildiği şekilde, hayvan kaynaklı dokular veya hücreler ya da bunların türevleri, e) Uygulanabildiği hallerde, bu Ekin 10.4.5 numaralı maddesi uyarınca etiketlenen bilgiler, f) “LOT NUMARASI” veya “SERİ NUMARASI” kelimelerinin ya da uygun olduğu takdirde eşdeğer bir sembolün ardından gelen cihazın lot numarası veya seri numarası, g) 27 nci maddenin dördüncü fıkrasında ve Ek VI Kısım C’de atıfta bulunulan UDI taşıyıcısı, ğ) İlgili olduğu durumda, cihazı güvenli bir şekilde kullanmaya veya implante etmeye yönelik süre sınırıyla ilgili, asgari yıl ve ay şeklinde ifade edilen açık bir gösterge, h) Cihazın güvenli bir şekilde hangi tarihe kadar kullanılabileceğine dair hiçbir göstergenin bulunmaması durumunda, imalat tarihi. Bu imalat tarihi, tarihin açıkça belirlenebilir olması şartıyla, lot numarasının veya seri numarasının bir parçası olarak dâhil edilebilir. ı) Özel depolama ve/veya kullanım koşullarına dair bir gösterge, i) Cihaz steril olarak tedarik ediliyorsa, cihazın steril durumuna ve sterilizasyon yöntemine dair bir gösterge, j) Cihaz kullanıcısının ve diğer kişilerin dikkatini hemen çekmesi gereken uyarılar veya alınacak önlemler. Bu bilgiler; kullanım talimatında daha detaylı bilgiler bulunduğu takdirde, hedeflenen kullanıcılar dikkate alınarak asgari düzeyde tutulabilir, k) Cihazın tek kullanımlık olması amaçlanmış ise bu durumla ilgili bir gösterge. İmalatçının tek kullanımlık göstergesi, Türkiye ve AB üyesi ülkeler genelinde tutarlı olur, l) Cihaz yeniden işlenmiş tek kullanımlık bir cihaz ise bu duruma dair bir gösterge, hâlihazırda yapılmış olan yeniden işleme döngülerinin sayısı ve yeniden işleme döngülerinin sayısına ilişkin sınırlamalar, m) Cihaz kişiye özel imal edilen bir cihaz ise “ısmarlama imal edilen cihazdır” ibaresi, n) Cihazın bir tıbbi cihaz olduğuna dair gösterge. Cihaz, sadece klinik araştırma amaçlı olarak imal edilmişse “sadece klinik araştırmaya mahsustur” ibaresi, o) Bir vücut açıklığı yoluyla insan vücuduna girmesi veya deriye uygulanması amaçlanan ve insan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar söz konusu olduğunda, cihazın genel niteliksel bileşimi ve hedeflenen asli etkiyi gerçekleştirmekten sorumlu ana bileşen veya bileşenleri hakkında niceliksel bilgiler, ö) İmplante edilebilir aktif cihazlar için seri numarası ve diğer implante edilebilir cihazlar için seri numarası veya lot numarası. 23.3. Bir cihazın steril durumunu muhafaza eden ambalaj (steril ambalaj) üzerindeki bilgiler Steril ambalaj üzerinde aşağıdaki ayrıntılar bulunur: a) Steril ambalajın tanınmasına imkân veren bir gösterge. b) Cihazın steril durumda olduğuna dair beyan. c) Sterilizasyon yöntemi. ç) İmalatçının adı ve adresi. d) Cihazın tanımı. e) Cihaz, sadece klinik araştırma amaçlı olarak imal edilmişse “sadece klinik araştırmaya mahsustur” ibaresi. f) Cihaz, kişiye özel imal edilen bir cihaz ise “ısmarlama imal edilen cihazdır” ibaresi. g) İmalat tarihi (ay ve yıl). h) Cihazı güvenli bir şekilde kullanmaya veya implante etmeye yönelik süre sınırıyla ilgili, en azından yıl ve ay şeklinde ifade edilen açık bir gösterge. ı) Steril ambalajın kullanım öncesinde hasar görmesi veya istenmeyen bir şekilde açılması halinde yapılması gerekenlere ilişkin olarak kullanım talimatının kontrol edilmesine yönelik bir talimat. 23.4. Kullanım talimatındaki bilgiler Kullanım talimatı, aşağıdakilerin tümünü içerir: a) Bu Ekin 23.2 numaralı maddesinin (a), (c), (d), (e), (ı), (i), (k) ve (o) bentlerinde atıfta bulunulan ayrıntılar, b) Uygun olduğu hallerde, endikasyonlar, kontrendikasyonlar, hedef hasta grubu veya grupları ve hedeflenen kullanıcılar hakkında açık bir tanımlama ile birlikte cihazın kullanım amacı, c) Uygulanabildiği hallerde, beklenen klinik faydalarla ilgili bir açıklama, ç) Uygulanabildiği hallerde, 32 nci maddede atıfta bulunulan güvenlilik ve klinik performans özeti için bağlantılar, d) Cihazın performans karakteristikleri, e) Uygulanabildiği hallerde, sağlık profesyonellerinin cihazın uygun olup olmadığını doğrulamasına ve ilgili yazılım ve aksesuarları seçmesine imkân veren bilgiler, f) Artık riskler, kontrendikasyonlar ve istenmeyen yan etkiler ile birlikte bu konuda hastaya iletilmesi gereken bilgiler, g) Kullanıcının cihazı uygun şekilde kullanması için gerekli olan açıklamalar; örneğin cihazın bir ölçüm fonksiyonu varsa buna yönelik iddia edilen doğruluk derecesi, ğ) Hasta güvenliğini sağlamak için gerekli olan dezenfeksiyon seviyeleri ve bu dezenfeksiyon seviyelerini gerçekleştirmeye yönelik mevcut tüm yöntemler dâhil olmak üzere, cihaz kullanıma hazır olmadan önce veya kullanım süresince cihazın hazırlık işlemleri veya kullanımı ile ilgili ayrıntılar (örneğin, sterilizasyon, nihai montaj, kalibrasyon ve benzeri), h) Özel tesislere ya da cihaz kullanıcısının ve/veya diğer kişilerin özel eğitimine ya da belirli yeterliliklerine yönelik gereklilikler, ı) İlgili olduğu yerde, aşağıdakilerle birlikte, cihazın doğru bir şekilde kurulup kurulmadığını ve güvenli bir şekilde ve imalatçı tarafından amaçlandığı gibi çalışmaya hazır olup olmadığını doğrulamak için gereken bilgiler: - Önleyici ve düzenli bakımın ve ön temizliğin veya dezenfeksiyonun niteliği ve sıklığına dair ayrıntılar, - Sarf malzeme bileşenlerinin ve bunların nasıl değiştirileceğinin tanımlanması, - Öngörülen kullanım ömrü boyunca cihazın doğru ve güvenli bir şekilde çalışmasını sağlamak için gerekli kalibrasyon bilgileri ve - Cihazların kurulumunda, kalibrasyonunda veya servis hizmetlerinde yer alan kişilerin karşılaştığı riskleri ortadan kaldırmaya yönelik yöntemler, i) Cihaz steril durumda tedarik ediliyor ise kullanım öncesinde steril ambalajının hasar görmesi veya istenmeyen bir şekilde açılması durumuna yönelik talimatlar, j) Cihaz, kullanım öncesinde steril edilmesi amacıyla steril olmayan durumda tedarik ediliyor ise sterilizasyona yönelik uygun talimatlar, k) Cihaz tekrar kullanılabilir ise temizlik, dezenfeksiyon, ambalajlama dâhil olmak üzere yeniden kullanımı mümkün kılmaya yönelik uygun süreçler ve yeniden sterilizasyona ilişkin geçerli kılınmış yöntem hakkında bilgiler. Malzeme bozulmasının belirtileri veya izin verilebilir azami yeniden kullanım sayısı gibi cihazın bundan sonra yeniden kullanılmaması gereken zamanı belirlemeye yönelik bilgiler sağlanır, l) Uygun olduğu hallerde, genel güvenlilik ve performans gerekliliklerine uymak için yalnızca imalatçının sorumluluğu altında rekondüksiyonu şartıyla cihazın tekrar kullanılabileceğine dair bir gösterge, m) Cihaz tek kullanımlık olduğuna dair bir gösterge taşıyor ise imalatçı tarafından bilinen, cihazın yeniden kullanılması halinde bir risk oluşturabilecek karakteristikler ve teknik faktörler hakkında bilgiler. Söz konusu bilgiler, bu tür karakteristiklerin ve teknik faktörlerin ayrıntılı olarak ele alındığı, imalatçının risk yönetimi dokümantasyonunun spesifik bir bölümüne dayanır. Bu Ekin 23.1 numaralı maddesinin (ç) bendi uyarınca kullanım talimatı gerekli değilse, bu bilgiler talebi halinde kullanıcıya sunulur, n) Diğer cihazlarla ve/veya genel amaçlı ekipmanla birlikte kullanılması amaçlanan cihazlar için: - Güvenli bir kombinasyon elde etmek üzere, bu tür cihazları veya ekipmanı tanımlamaya yönelik bilgiler ve/veya - Cihaz ve ekipman kombinasyonlarına dair bilinen kısıtlamalara ilişkin bilgiler, o) Cihazın tıbbi amaçlar için radyasyon yayması halinde: - Yayılan radyasyonun niteliğine, türüne ve uygun olduğu hallerde yoğunluğuna ve dağılımına ilişkin ayrıntılı bilgiler, - Cihazın kullanımı süresince hastayı, kullanıcıyı veya diğer kişiyi istenmeyen radyasyondan koruma araçları, ö) Kullanıcının ve/veya hastanın, uyarılar, önlemler, kontrendikasyonlar, alınacak tedbirler ve cihaza ilişkin kullanım sınırlamaları hakkında bilgilendirilmesine imkân veren bilgiler. Bu bilgiler; ilgili olduğu durumda, uyarılar, önlemler, kontrendikasyonlar, alınacak tedbirler ve cihaza ilişkin kullanım sınırlamaları hakkında kullanıcının hastayı bilgilendirmesine olanak sağlar. Bu bilgiler, uygun olduğu hallerde aşağıdakileri kapsar: - Cihaz arızası ya da cihazın performansında güvenliği etkileyebilecek değişiklikler meydana gelmesi durumunda uyarılar, önlemler ve/veya alınacak tedbirler, - Manyetik alanlar, harici elektriksel ve elektromanyetik etkiler, elektrostatik boşalma, diyagnostik veya terapötik prosedürler ile ilişkili radyasyon, basınç, nem veya sıcaklık gibi makul olarak öngörülebilir dış etkilere veya çevresel şartlara maruz kalmayla ilgili uyarılar, önlemler ve/veya alınacak tedbirler, - Diyagnostik amaçlı spesifik araştırmalar, değerlendirmeler veya terapötik amaçlı işlemler ya da diğer prosedürler sırasında, makul ve öngörülebilir bir şekilde cihazın mevcudiyetinden kaynaklanan girişim riskleri (örneğin, cihazdan yayılan ve diğer ekipmanı etkileyen elektromanyetik interferans) ile ilgili uyarılar, önlemler ve/veya alınacak tedbirler, - Cihaz; tıbbi ürünleri, insan veya hayvan kaynaklı dokuları ya da hücreleri veya bunların türevlerini ya da biyolojik maddeleri tatbik etme amaçlı ise verilecek maddelerin seçimindeki sınırlamalar veya uyumsuzluklar, - Cihazın bütünleşik bir parçası olarak ihtiva ettiği tıbbi madde veya biyolojik materyalle ilgili uyarılar, önlemler ve/veya sınırlamalar ve - Cihazın ihtiva ettiği, CMR maddeler veya endokrin-bozucu maddeler içeren veya bunlardan oluşan ya da hastada veya kullanıcıda hassasiyete veya alerjik bir reaksiyona yol açabilecek olan materyallerle ilgili önlemler, p) İnsan vücuduna girmesi amaçlanan ve insan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar söz konusu olduğunda; uygun olduğu hallerde, kontrendikasyonlar, istenmeyen yan etkiler ve doz aşımı ile ilgili risklerin yanı sıra cihazın ve onun metabolizma ürünlerinin diğer cihazlarla, tıbbi ürünlerle ve diğer maddelerle etkileşiminin genel profiliyle ilgili uyarılar ve önlemler, r) İmplante edilebilir cihazlar söz konusu olduğunda; hastaların maruz kalabileceği materyaller ve maddeler hakkında tüm niteliksel ve niceliksel bilgiler, s) Cihazın, varsa aksesuarlarının ve cihazla birlikte kullanılan sarf malzemelerin güvenli bir şekilde bertarafını kolaylaştırmak için uyarılar veya alınacak önlemler. Bu bilgiler, uygun olduğu hallerde aşağıdakileri kapsar: - Enfeksiyon veya mikrobiyal tehlikeler (örneğin, insan kaynaklı potansiyel olarak enfeksiyöz maddelerle kontamine olan eksplantlar, iğneler veya cerrahi ekipman) ve - Keskin aletlerden kaynaklananlar gibi fiziksel tehlikeler. Bu Ekin 23.1. numaralı maddesinin (ç) bendi uyarınca kullanım talimatı gerekli değilse, bu bilgiler talebi halinde kullanıcıya sunulur, ş) Meslekten olmayan kişiler tarafından kullanılması amaçlanan cihazlar için kullanıcının bir sağlık profesyoneline danışması gereken durumlar, t) 1 inci maddenin üçüncü fıkrasının (b) bendi uyarınca bu Yönetmeliğin kapsadığı cihazlar için klinik bir faydanın olmadığına ilişkin bilgiler ve cihazın kullanımıyla ilgili riskler, u) Kullanım talimatının basım tarihi veya revize edilmiş ise en son revizyonunun tanımlayıcısı ve basım tarihi, ü) Cihazla ilgili meydana gelen herhangi bir ciddi olumsuz olayın imalatçıya ve Kuruma raporlanması gerektiğine ilişkin kullanıcıya ve/veya hastaya yönelik bir bildirim, v) İmplante edilmiş bir cihazı olan hastaya 18 inci madde uyarınca sağlanacak bilgiler, y) Yazılım dâhil olmak üzere elektronik programlanabilir sistemler içeren cihazlar ya da kendi başına cihaz olan yazılımlar için; yazılımın amaçlandığı şekilde çalışması için gerekli olan, donanım, BT ağ karakteristikleri ve yetkisiz erişime karşı koruma dâhil BT güvenlik önlemleri ile ilgili asgari gereklilikler.
______________________________________
Ek II TEKNİK DOKÜMANTASYON İmalatçı tarafından hazırlanacak teknik dokümantasyon ve uygulanabildiği hallerde bunun özeti; açık, düzenli, kolaylıkla incelenebilir ve belirsizliğe mahal vermeyecek bir şekilde sunulur ve bu Ekte listelenen unsurları özellikle içerir. 1. VARYANTLARI VE AKSESUARLARI DÂHİL OLMAK ÜZERE CİHAZ TANIMI VE SPESİFİKASYONLARI 1.1. Cihaz tanımı ve spesifikasyonları a) Ürünün adı veya ticari adı ve kullanım amacı ile hedeflenen kullanıcılar dâhil olmak üzere cihazın genel bir tanımı, b) Cihazın tanımlanması UDI sistemine dayalı hale gelir gelmez söz konusu cihaz için imalatçı tarafından tahsis edilen, Ek VI Kısım C’de atıfta bulunulduğu üzere Temel UDI-DI ya da aksi takdirde ürün kodu, katalog numarası veya izlenebilirliği mümkün kılan başka bir özgün atıf yoluyla açık bir tanımlama, c) Hedeflenen hasta popülasyonu ve tanılanacak, tedavi edilecek ve/veya izlenecek tıbbi durumlar ve hasta seçim kriterleri, endikasyonlar, kontrendikasyonlar, uyarılar gibi diğer hususlar, ç) Gerektiğinde bilimsel olarak gösterilmiş, cihazın çalışma ilkeleri ve etki mekanizması, d) Ürünün bir cihaz olarak nitelendirilmesine ilişkin gerekçe, e) Cihazın risk sınıfı ve Ek VIII uyarınca uygulanan sınıflandırma kural/kurallarına ilişkin gerekçe, f) Her türlü yeni özellik ile ilgili açıklama, g) Cihazla birlikte kullanılması amaçlanan cihaz aksesuarlarının, diğer cihazların ve cihaz olmayan diğer ürünlerin tanımı, ğ) Piyasada bulundurulması amaçlanan cihazın çeşitli konfigürasyonlarının/ varyantlarının tanımı veya tam listesi, h) Parçalar/bileşenler (uygun olduğu hallerde yazılım dâhil), formülasyon, bileşim, işlevsellik ve ilgili olduğu yerde niteliksel ve niceliksel bileşim gibi temel işlevsel ögelerin genel bir tanımı. Uygun olduğu hallerde bu tanım; temel parçaları/bileşenleri açıkça gösteren etiketlenmiş resimli anlatımlar (örneğin, diyagramlar, fotoğraflar ve çizimler) ile bu çizimlerin ve diyagramların anlaşılmasına yönelik yeterli açıklama içerir, ı) Temel işlevsel ögelere dâhil edilen hammaddelerin ve insan vücuduna doğrudan veya dolaylı olarak temas eden hammaddelerin (örneğin, vücut sıvılarının ekstrakorporeal dolaşımı süresince olanların) tanımı, i) Broşürlerde, kataloglarda ve benzer yayınlarda kullanıcıya sunulan ürün özelliklerinde tipik olarak yer alan, cihazın ve tüm varyantlarının/konfigürasyonlarının ve aksesuarlarının özellikler, boyutlar ve performans nitelikleri gibi teknik spesifikasyonları. 1.2. Cihazın önceki ve benzer nesillerine atıf a) İmalatçı tarafından üretilen cihazın, varsa önceki nesli veya nesilleri hakkında genel bir açıklama, b) Varsa, yurt içi piyasada veya uluslararası piyasalarda bulunan, benzer olarak tanımlanmış cihazlar hakkında genel bir açıklama. 2. İMALATÇI TARAFINDAN SAĞLANAN BİLGİLER Aşağıdakilerden oluşan tam bir bilgi seti: - 7223 sayılı Kanunun 7 nci maddesinin birinci fıkrasının (ğ) bendine uygun olarak cihazın ve ambalajının (örneğin, tekli birim ambalajı, satış ambalajı, spesifik sevk ve idare şartlarında nakliye ambalajı) üzerindeki etiket veya etiketler ve kullanım talimatları. 3. TASARIM VE İMALAT BİLGİLERİ a) Cihaza uygulanan tasarım aşamalarının anlaşılmasına olanak sağlayacak bilgiler, b) İmalat süreçleri ve bunların validasyonu, destekleyicileri, sürekli izleme ve nihai ürün testi dâhil olmak üzere tüm bilgi ve spesifikasyonlar. Veriler, teknik dokümantasyona tümüyle dâhil edilir, c) Tedarikçiler ve yükleniciler dâhil olmak üzere, tasarım ve imalat faaliyetlerinin yürütüldüğü tüm tesislerin tanımlanması. 4. GENEL GÜVENLİLİK VE PERFORMANS GEREKLİLİKLERİ Dokümantasyon; kullanım amacı dikkate alınarak cihaza uygulanabilir olan ve Ek I’de belirtilen genel güvenlilik ve performans gerekliliklerine uygunluğun gösterilmesine yönelik bilgileri kapsar ve bu gereklilikleri karşılamak için kabul edilen çözümlerin gerekçesini, validasyonunu ve doğrulamasını içerir. Uygunluğun gösterilmesi aşağıdakileri içerir: a) Cihaza uygulanan genel güvenlilik ve performans gereklilikleri ve diğerlerinin neden uygulanmadığına ilişkin bir açıklama, b) Uygulanabilir her bir genel güvenlilik ve performans gerekliliğine uygunluğu göstermek için kullanılan metot/metotlar, c) Uygulanan uyumlaştırılmış standartlar, ortak spesifikasyonlar veya diğer çözümler ve ç) Genel güvenlilik ve performans gerekliliklerine uygunluğu göstermek amacıyla uygulanan her bir uyumlaştırılmış standarda, ortak spesifikasyona veya diğer metoda uygunluğun kanıtı olarak sunulan kontrollü belgelerin kesin tanımı. Burada belirtilen bilgiler; bu tür kanıtın teknik dokümantasyonda ve uygulanabildiği hallerde teknik dokümantasyonun özetinde nerede bulunduğuna dair bir atıf içerir. 5. FAYDA-RİSK ANALİZİ VE RİSK YÖNETİMİ Dokümantasyon aşağıdakilere ilişkin bilgiler içerir: a) Ek I’in 1 ve 8 numaralı maddelerinde atıfta bulunulan fayda-risk analizi ve b) Benimsenen çözümler ve Ek I’in 3 numaralı maddesinde atıfta bulunulan risk yönetimiyle ilgili sonuçlar. 6. ÜRÜN DOĞRULAMA VE VALİDASYON Dokümantasyon; cihazın bu Yönetmeliğin gerekliliklerine ve özellikle uygulanabilir genel güvenlilik ve performans gerekliliklerine uygunluğunu göstermek için yürütülen tüm doğrulamalar ve validasyon testleri ve/veya çalışmalar ile ilgili sonuçları ve kritik analizleri içerir. 6.1. Klinik-öncesi ve klinik veriler a) Kullanım amacını dikkate alarak, cihazın klinik öncesi güvenliğiyle ve spesifikasyonlara uygunluğuyla ilgili olarak; mühendislik, laboratuvar, simüle edilmiş kullanım ve hayvan testleri gibi testlerin sonuçları ve cihaza ya da benzer cihazlara uygulanabilir yayımlanmış literatürün değerlendirmesi, b) Özellikle aşağıdakilerle ilgili veri özetlerine ve test sonuçlarına ilave olarak; test tasarımına, tüm test veya çalışma protokollerine, veri analizi yöntemlerine ilişkin ayrıntılı bilgiler: - Hasta veya kullanıcıya doğrudan veya dolaylı olarak temas eden bütün malzemelerin tanımlanması dâhil cihazın biyouyumluluğu, - Fiziksel, kimyasal ve mikrobiyolojik karakterizasyon, - Elektriksel güvenlik ve elektromanyetik uyumluluk, - Yazılım doğrulama ve validasyon (Yazılım tasarım ve geliştirme sürecini tanımlar ve bitmiş cihazda kullanıldığı şekilde, yazılım validasyonu ile ilgili kanıtları gösterir. Bu bilgiler, genellikle nihai sürüm öncesinde, hem kuruluş bünyesinde hem de simüle edilmiş veya gerçek bir kullanıcı ortamında gerçekleştirilmiş bütün doğrulama, validasyon ve testlerin özet sonuçlarını içerir. Bu bilgiler, farklı donanım konfigürasyonlarının ve uygulanabildiği hallerde, imalatçı tarafından temin edilen bilgilerde tanımlanmış işletim sistemlerinin tamamını da ele alır), - Raf ömrü dâhil stabilite ve - Performans ve güvenlilik. Uygulanabildiği hallerde, 9/3/2010 tarihli ve 27516 sayılı Resmî Gazete’de yayımlanan İyi Laboratuvar Uygulamaları Prensipleri, Test Birimlerinin Uyumlaştırılması, İyi Laboratuvar Uygulamalarının ve Çalışmaların Denetlenmesi Hakkında Yönetmelik hükümlerine uygunluk gösterilir. Yeni bir test yapılmamışsa, dokümantasyon bu karar için bir gerekçe içerir. Yasal olarak piyasaya arz edilmiş veya hizmete sunulmuş olan cihazın önceki bir versiyonu, özdeş malzemeleri ihtiva ettiğinde, bu malzemeler ile ilgili biyouyumluluk testinin yapılmış olması bu tür bir gerekçeye örnek olabilir. c) 61 inci maddenin on birinci fıkrasında ve Ek XIV’ün A Kısmında atıfta bulunulan klinik değerlendirme raporu ve güncellemeleri ile klinik değerlendirme planı, ç) Ek XIV Kısım B’de atıfta bulunulan piyasaya arz sonrası klinik takip (PMCF) planı ve piyasaya arz sonrası klinik takip planı değerlendirme raporu ya da piyasaya arz sonrası klinik takip planının neden uygulanabilir olmadığına dair bir gerekçe. 6.2. Özel durumlarda gereken ilave bilgiler a) Bir cihazın, bütünleşik bir parça olarak, 1 inci maddenin altıncı fıkrasının ikinci cümlesinde atıfta bulunulduğu şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün dâhil, ayrı olarak kullanıldığında Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin 4 üncü maddesinin (b) bendi çerçevesinde tıbbi ürün olarak kabul edilebilen bir madde içermesi durumunda, bu durumu belirten bir beyan. Bu durumda, dokümantasyon; bu maddenin kaynağını tanımlar ve cihazın kullanım amacını dikkate alarak bu maddenin güvenliliğini, kalitesini ve yararlılığını değerlendirmek üzere yürütülen testlerle ilgili veriler içerir. b) Bir cihazın, insan veya hayvan kaynaklı dokular veya hücreler veya bunların türevleri kullanılarak imal edilmesi ve 1 inci maddenin dördüncü fıkrasının (e) ve (f) bentleri uyarınca bu Yönetmelik kapsamında olması halinde ve bir cihazın, bütünleşik bir parça olarak, cihazın fonksiyonuna yardımcı olan insan kaynaklı dokular veya hücreler ya da onların türevlerini ihtiva etmesi ve 1 inci maddenin sekizinci fıkrasının bir ve ikinci cümlesi uyarınca bu Yönetmeliğin kapsamında olması halinde, bu durumu belirten bir beyan. Bu durumda, dokümantasyon, kullanılan insan veya hayvan kaynaklı bütün materyalleri tanımlar ve Ek I’in sırasıyla, 13.1 veya 13.2 numaralı maddelerine uygunluğuna ilişkin ayrıntılı bilgiler sağlar. c) İnsan vücuduna girmesi amaçlanan ve insan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar söz konusu olduğunda; test tasarımı, tam test veya çalışma protokolleri, veri analiz yöntemleri ve veri özetleri ile test sonuçları dâhil, aşağıdakilerle ilgili çalışmalara ilişkin ayrıntılı bilgiler: - Absorbsiyon, dağılım, metabolizma ve eliminasyon, - Hedef popülasyonu ve hedef popülasyonun ilişkili tıbbi durumlarını göz önünde bulundurarak, bu maddelerin veya bunların insan vücudundaki metabolizma ürünlerinin; diğer cihazlar, tıbbi ürünler veya diğer maddeler ile olası etkileşimleri, - Lokal tolerans ve - Cihaza maruziyet düzeyine ve tipine bağlı olarak uygulanabildiği şekilde, tek-doz toksisitesi, yinelenen-doz toksisitesi, genotoksisite, karsinojenisite ile üreme ve gelişimsel toksisite dâhil, toksisite. Bu tür çalışmaların bulunmaması durumunda bir gerekçe sunulur. ç) Ek I’in 10.4.1 numaralı maddesinde atıfta bulunulan CMR veya endokrin-bozucu maddeler içeren cihazlar söz konusu olduğunda, bahsi geçen Ek’in 10.4.2 numaralı maddesinde atıfta bulunulan gerekçe. d) Steril veya tanımlanmış mikrobiyolojik bir durumda piyasaya arz edilen cihazlar söz konusu olduğunda; ilgili imalat adımlarına yönelik çevresel koşulların bir açıklaması. Steril olarak piyasaya arz edilen cihazlar söz konusu olduğunda; validasyon raporları dâhil olmak üzere ambalajlama, sterilizasyon ve sterilitenin muhafazası ile ilgili kullanılan yöntemlerin tanımı. Validasyon raporu, biyolojik yük (bioburden) testini, pirojen testini ve mevcutsa sterilant kalıntı testini ele alır. e) Ölçüm fonksiyonu ile piyasaya arz edilen cihazlar söz konusu olduğunda; spesifikasyonlarında verildiği şekilde doğruluğu sağlamak için kullanılan yöntemlerin tanımı. f) Cihaz amaçlandığı şekilde çalışması için başka bir cihaza veya cihazlara bağlanacak ise, imalatçı tarafından belirtilen karakteristikleri göz önüne alınarak bu tür cihaza veya cihazlara bağlandığında genel güvenlilik ve performans gerekliliklerine uyduğuna ilişkin kanıt dâhil, bu kombinasyon/ konfigürasyonun tanımı.
_______________________________________
Ek III PİYASAYA ARZ SONRASI GÖZETİME İLİŞKİN TEKNİK DOKÜMANTASYON 81 ila 84 üncü maddeler uyarınca imalatçı tarafından hazırlanacak piyasaya arz sonrası gözetime ilişkin teknik dokümantasyon; açık, düzenli, kolaylıkla incelenebilir ve belirsizliğe mahal vermeyecek bir şekilde sunulur ve özellikle bu Ekte belirtilen unsurları içerir. 1. 82 nci madde uyarınca hazırlanan piyasaya arz sonrası gözetim planı; İmalatçı; 81 inci maddede atıfta bulunulan yükümlülüğe uyduğunu piyasaya arz sonrası gözetim planında kanıtlar. a) Piyasaya arz sonrası gözetim planı, özellikle aşağıdaki mevcut bilgilerin toplanmasını ve kullanımını ele alır: - Periyodik güvenlilik güncelleme raporlarından alınan bilgiler dâhil ciddi olumsuz olaylara ve saha güvenliği düzeltici faaliyetlere ilişkin bilgiler, - Ciddi olmayan olumsuz olaylara atıfta bulunan kayıtlar ve istenmeyen yan etkilere ilişkin veriler, - Trend raporlamasından elde edilen bilgiler, - İlgili uzmanlık literatürü ya da teknik literatür, veri tabanları ve/veya kayıtlar, - Geri bildirimler ve şikayetler dâhil, kullanıcılar, dağıtıcılar ve ithalatçılar tarafından sağlanan bilgiler ve - Benzer tıbbi cihazlar hakkında kamuya açık bilgiler. b) Piyasaya arz sonrası gözetim planı, asgari olarak aşağıdakileri kapsar: - (a) bendinde atıfta bulunulan bilgileri toplamak üzere proaktif ve sistematik bir süreci. Bu süreç, cihazların performansıyla ilgili doğru bir karakterizasyona olanak sağlar ve ayrıca cihaz ile piyasada bulunan benzer ürünler arasında yapılacak bir karşılaştırmaya imkân tanır. - Toplanmış verileri değerlendirmek üzere etkili ve uygun yöntemleri ve süreçleri, - Fayda-risk analizinin ve Ek I’in 3 numaralı maddesinde atıfta bulunulduğu şekilde risk yönetiminin sürekli yeniden değerlendirilmesinde kullanılan uygun indikatörleri ve eşik değerleri, - Şikâyetleri araştırmak ve sahadan toplanan piyasayla ilgili deneyimleri analiz etmek üzere etkili ve uygun yöntemleri ve araçları, - Gözlem periyoduyla birlikte, olumsuz olayların sıklığındaki veya şiddetindeki istatistiksel olarak anlamlı artışları belirlemek için kullanılacak yöntemler ve protokoller dâhil, 86 ncı maddede belirtildiği şekilde trend raporuna tabi olan olumsuz olayları yönetmek üzere yöntemleri ve protokolleri, - Yetkili otoriteler, onaylanmış kuruluşlar, iktisadi işletmeciler ve kullanıcılarla etkili iletişim için yöntemleri ve protokolleri, - 81, 82 ve 84 üncü maddelerde belirtilen imalatçı yükümlülüklerini yerine getirmeye yönelik prosedürlere atfı, - Düzeltici faaliyetler dâhil, uygun önlemleri tanımlamak ve başlatmak için sistematik prosedürleri, - Düzeltici faaliyet gerektirebilecek cihazları izlemek ve tanımlamak için etkili araçları ve - Ek XIV Kısım B’de atıfta bulunulduğu şekilde bir piyasaya arz sonrası klinik takip planını ya da piyasaya arz sonrası klinik takibin neden uygulanabilir olmadığına dair bir gerekçeyi. 2. 84 üncü maddede atıfta bulunulan periyodik güvenlilik güncelleme raporu ve 83 üncü maddede atıfta bulunulan piyasaya arz sonrası gözetim raporu.
__________________________________________
Ek IV AB UYGUNLUK BEYANI AB uygunluk beyanı aşağıdaki bilgilerin tamamını içerir: 1. İmalatçının ve mevcutsa yetkili temsilcisinin adı, kayıtlı ticari unvanı veya kayıtlı ticari markası ve hâlihazırda düzenlenmişse, 31 inci maddede atıfta bulunulduğu şekilde münferit kimlik numarası ve kendilerine ulaşılabilecek kayıtlı iş yerlerinin adresi; 2. İmalatçının AB uygunluk beyanını tamamen kendi sorumluluğunda düzenlediğine ilişkin bir ifade, 3. Ek VI Kısım C’de atıfta bulunulduğu şekilde Temel UDI-DI, 4. AB uygunluk beyanı kapsamındaki cihazın, tanımlanmasına ve izlenebilirliğine imkan verecek şekilde, ürün adı ve ticari adı, ürün kodu, katalog numarası veya uygulanabilir olduğu hallerde fotoğraf gibi diğer kesin referanslar ile birlikte kullanım amacı. Ürün adı veya ticari adı hariç olmak üzere, tanımlamaya ve izlenebilirliğe olanak sağlayan bilgiler bu Ekin 3 numaralı maddesinde atıfta bulunulan Temel UDI-DI ile sağlanabilir. 5. Ek VIII’de belirtilen kurallar uyarınca cihazın risk sınıfı, 6. Mevcut beyan kapsamında olan cihazın bu Yönetmeliğe ve uygulanabilirse, bir AB uygunluk beyanının düzenlenmesini şart koşan ilgili diğer AB mevzuatına uygun olduğuna dair bir ifade, 7. Kullanılan ve uygunluğun beyan edildiği ortak spesifikasyonlara ilişkin atıflar, 8. Uygulanabildiği hallerde, onaylanmış kuruluşun adı ve kimlik numarası, yürütülen uygunluk değerlendirme prosedürünün bir tanımı ve düzenlenen sertifika veya sertifikaların tanımlanması, 9. Uygulanabildiği hallerde ilave bilgiler, 10. Beyanın düzenlenme yeri ve tarihi, imzalayan kişinin adı ve görevi ile birlikte bu kişinin kimin adına imzaladığının bilgisi, imza.
__________________________________________
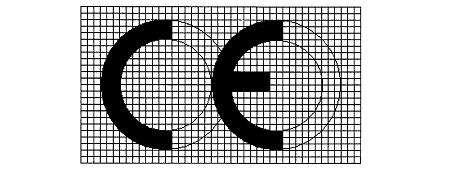
Ek V CE UYGUNLUK İŞARETİ 1. CE işareti, aşağıdaki şekli alan “CE” harflerinden oluşur:
2. CE işareti küçültülür veya büyütülür ise yukarıda yer alan ölçekli çizimde belirtilen oranlara uyulur. 3. CE işaretinin bileşenleri esasen 5 mm’den az olmayan aynı dikey boyuta sahip olur. Bu asgari boyut, küçük boyutlu cihazlar için uygulanmayabilir.
______________________________________
Ek VI CİHAZLARIN VE İKTİSADİ İŞLETMECİLERİN KAYDI İÇİN SUNULACAK BİLGİLER, UDI-DI İLE BİRLİKTE UDI VERİ TABANINA SAĞLANACAK TEMEL VERİ ÖGELERİ ve UDI SİSTEMİ
KISIM A 29 UNCU MADDENİN DÖRDÜNCÜ FIKRASI ve 31 İNCİ MADDE UYARINCA CİHAZLARIN ve İKTİSADİ İŞLETMECİLERİN KAYDI İÇİN SUNULACAK BİLGİLER İmalatçılar ya da uygun olduğu hallerde, yetkili temsilciler ve ithalatçılar; bu Kısmın 1. numaralı maddesinde atıfta bulunulan bilgileri sunar ve bu Kısmın 2. numaralı maddesinde atıfta bulunulan bilgilerin tam, doğru ve ilgili tarafça güncellenmiş olmasını sağlar. 1. İktisadi işletmeciyle ilgili bilgiler 1.1. İktisadi işletmecinin türü (imalatçı, yetkili temsilci veya ithalatçı), 1.2. İktisadi işletmecinin adı, adresi ve iletişim bilgileri, 1.3. Bilgilerin sunumunun bu Kısmın 1.1. numaralı maddesinde bahsedilen iktisadi işletmecilerden herhangi biri adına başka bir kişi tarafından yerine getirilmesi durumunda, ilgili kişinin adı, adresi ve iletişim bilgileri, 1.4. 15 inci maddede atıfta bulunulan mevzuata uyum sorumlusu veya sorumlularının adı, adresi ve iletişim bilgileri. 2. Cihazla ilgili bilgiler 2.1. Temel UDI-DI, 2.2. Onaylanmış kuruluş tarafından düzenlenen sertifikanın tipi, numarası ve bitiş tarihi, bu onaylanmış kuruluşun adı veya kimlik numarası ile sertifikada yer alan ve onaylanmış kuruluşlara ve sertifikalara ilişkin elektronik sisteme onaylanmış kuruluş tarafından girilen bilgilere yönelik bağlantı, 2.3. Cihazın piyasaya arz edileceği veya arz edilmiş olduğu Türkiye / AB üyesi ülkeler, 2.4. Sınıf IIa, sınıf IIb veya sınıf III cihazlar söz konusu olduğunda; cihazın bulundurulduğu veya bulundurulacağı Türkiye / AB üyesi ülkeler, 2.5. Cihazın risk sınıfı, 2.6. Yeniden işlenmiş tek kullanımlık cihaz (evet/hayır), 2.7. Ayrı olarak kullanıldığı takdirde tıbbi ürün olarak kabul edilebilen bir maddenin mevcudiyeti ve bu maddenin adı, 2.8. Ayrı olarak kullanıldığında insan kanından veya insan plazmasından elde edilen bir tıbbi ürün olarak kabul edilebilen bir maddenin mevcudiyeti ve bu maddenin adı, 2.9. İnsan kaynaklı dokuların veya hücrelerin ya da türevlerinin mevcudiyeti (evet/hayır), 2.10. Hayvan Kaynaklı Dokular Kullanılarak İmal Edilen Tıbbi Cihazlara Dair Yönetmelikte atıfta bulunulduğu şekilde, hayvan kaynaklı dokuların veya hücrelerin ya da türevlerinin mevcudiyeti (evet/hayır), 2.11. Uygulanabildiği hallerde, cihazla ilgili olarak yürütülen klinik araştırma veya araştırmalara özgü tek bir kimlik numarasına veya klinik araştırmalara yönelik elektronik sistemdeki klinik araştırma kaydına bir bağlantı, 2.12. Ek XVI’da listelenen cihazlar söz konusu olduğunda, cihazın kullanım amacının tıbbi bir amaç dışında olduğuna ilişkin açıklama, 2.13. 10 uncu maddenin on beşinci fıkrasında atıfta bulunulduğu şekilde, başka bir tüzel veya gerçek kişi tarafından tasarlanan ve imal edilen cihazlar söz konusu olduğunda, ilgili tüzel veya gerçek kişinin adı, adresi ve iletişim bilgileri, 2.14. Sınıf III veya implante edilebilir cihazlar söz konusu olduğunda, güvenlilik ve klinik performans özeti, 2.15. Cihazın durumu (piyasada/ artık piyasaya arz edilmiyor/ geri çağrılmış/ saha güvenliği düzeltici faaliyeti başlatılmış).
KISIM B 28 İNCİ ve 29 UNCU MADDELER UYARINCA UDI-DI İLE BİRLİKTE UDI VERİ TABANINA SAĞLANACAK TEMEL VERİ ÖGELERİ İmalatçı, UDI veri tabanına, UDI-DI ile birlikte imalatçı ve cihazla ilgili aşağıdaki bilgilerin tamamını girer: 1. Ambalaj konfigürasyonu başına düşen miktar, 2. 29 uncu maddede atıfta bulunulduğu şekilde Temel UDI-DI ve herhangi ilave UDI-DI’lar, 3. Cihaz üretiminin kontrol edilme şekli (son kullanma tarihi veya imalat tarihi, lot numarası, seri numarası) 4. Mevcutsa, UDI-DI kullanım birimi (bir UDI’nın, cihazın üzerinde kullanım birimi seviyesinde etiketlenmemesi durumunda, cihazın kullanımını hasta ile ilişkilendirmek için bir “kullanım birimi” DI, atanır), 5. İmalatçının adı ve adresi (etiket üzerinde belirtildiği şekilde), 6. 31 inci maddenin ikinci fıkrası uyarınca verilen münferit kayıt numarası (SRN), 7. Mevcutsa, yetkili temsilcinin adı ve adresi (etikette belirtildiği şekilde), 8. 26 ncı maddede belirtildiği şekilde tıbbi cihaz terminolojisi kodu, 9. Cihazın risk sınıfı, 10. Cihazın adı veya ticari adı, 11. Cihaz modeli ya da referans veya katalog numarası 12. Mevcutsa, hacim, uzunluk, ölçek, çap dâhil klinik boyut, 13. Ek ürün açıklaması (isteğe bağlı), 14. Uygulanabilirse depolama ve/veya kullanım şartları (etiket üzerinde veya kullanım talimatında belirtildiği şekilde), 15. Uygulanabilirse, cihazın ilave ticari adları, 16. Tek kullanımlık cihaz olarak etiketlenmiş (evet/hayır), 17. Uygulanabilirse, azami yeniden kullanım sayısı, 18. Steril etiketli cihaz (evet/hayır), 19. Kullanımdan önce sterilizasyon ihtiyacı (evet/hayır), 20. Lateks içeriyor (evet/hayır) 21. Uygulanabildiği hallerde, Ek I’in 10.4.5 numaralı maddesi uyarınca etiketlenen bilgiler, 22. Elektronik kullanım talimatı gibi ilave bilgiler için URL (isteğe bağlı), 23. Mevcutsa, kritik uyarılar veya kontrendikasyonlar, 24. Cihazın durumu (piyasada/artık piyasaya arz edilmiyor/geri çağrılmış/saha güvenliği düzeltici faaliyeti başlatılmış)
KISIM C UDI SİSTEMİ 1. Tanımlar 1.1. Otomatik tanımlama ve veri toplama (AIDC) AIDC, otomatik olarak veri toplamak için kullanılan bir teknolojidir. AIDC teknolojileri; barkodları, akıllı kartları, biyometrikleri ve RFID’yi içerir. 1.2. Temel UDI-DI Temel UDI-DI, bir cihaz modelinin birincil tanımlayıcısıdır. Cihazın kullanım birimi seviyesinde atanan DI’sıdır. UDI veri tabanına kayıtların ana anahtarıdır ve ilgili sertifikalarda ve AB uygunluk beyanlarında atıfta bulunulur. 1.3. Kullanım birimi DI Kullanım Birimi DI, bir UDI’nin, kullanım birimi seviyesinde tek bir cihazın üzerinde etiketlenmediği örneğin, aynı cihazın birden fazla biriminin birlikte ambalajlandığı durumlarda, cihaz kullanımını hasta ile ilişkilendirmeye hizmet eder. 1.4. Konfigüre edilebilir cihaz Konfigüre edilebilir cihaz; imalatçı tarafından birden fazla konfigürasyonda birleştirilebilen birkaç bileşenden oluşan cihazdır. Bu münferit bileşenler, kendi başlarına cihazlar olabilir. Konfigüre edilebilir cihazlar, bilgisayarlı tomografi sistemlerini, ultrason sistemlerini, anestezi sistemlerini, fizyolojik izleme sistemlerini ve radyoloji bilgi sistemlerini içerir. 1.5. Konfigürasyon Konfigürasyon; imalatçı tarafından belirtildiği şekilde, kullanım amacını gerçekleştirmek için bir cihaz olarak birlikte çalışan ekipman parçalarının kombinasyonudur. Parçaların kombinasyonu, belirli ihtiyaçları karşılamak için modifiye edilebilir, ayarlanabilir veya özelleştirilebilir. Konfigürasyonlar diğerlerine ilaveten aşağıdakileri içerir: - Bilgisayarlı tomografide amaçlanan bir fonksiyonu yerine getirmesi için konfigüre/kombine edilebilen gantriler, tüpler, masalar, konsollar ve diğer ekipman parçaları. - Anestezide amaçlanan bir fonksiyonu yerine getirmesi için kombine edilen ventilatörler, solunum devreleri, vaporizatörler. 1.6. UDI-DI UDI-DI; bir cihaz modeline spesifik olan tekil bir numerik veya alfanumerik koddur ve ayrıca UDI veri tabanında kayıtlı bilgilere “erişim anahtarı” olarak kullanılır. 1.7. İnsan tarafından okunabilir çeviri (HRI) HRI; UDI taşıyıcısında şifrelenmiş veri karakterlerinin okunabilir bir çevirisidir. 1.8. Ambalaj seviyeleri Ambalaj seviyeleri; tanımlanmış bir miktarda cihaz içeren, örneğin koli veya kutu gibi, cihaz ambalajının çeşitli seviyeleri anlamına gelir. 1.9. UDI-PI UDI-PI, üretim birimini tanımlayan bir numerik veya alfa numerik koddur. UDI-PI’ların farklı türleri; seri numarasını, lot numarasını, yazılım kimliğini ve imalat veya son kullanma tarihini ya da her iki tarih tipini içerir. 1.10. Radyo frekansı ile tanımlama RFID RFID; tanımlama amacıyla, bir okuyucu ile bir nesneye iliştirilen elektronik etiket arasında radyo dalgalarının kullanımı yoluyla veri alışverişine yönelik haberleşmeyi kullanan bir teknolojidir. 1.11. Nakliyat konteynırları Nakliyat konteynırı; lojistik sistemlere özel bir süreçle izlenebilirliğinin kontrol edildiği bir konteynırdır. 1.12. Tekil cihaz kimliği (UDI) UDI; küresel kabul görmüş bir cihaz tanımlama ve kodlama standardı vasıtasıyla oluşturulan bir numerik veya alfanumerik karakterler dizisidir. Piyasadaki belirli bir cihazın, belirsizliğe mahal vermeyecek şekilde tanımlanmasına olanak sağlar. UDI; UDI-DI ve UDI-PI’dan oluşur. ‘Tekil’ kelimesi, her bir üretim biriminin serileştirilmesi anlamına gelmez. 1.13. UDI taşıyıcısı UDI taşıyıcısı; UDI’yi AIDC ve mevcutsa, onun HRI’sını kullanarak iletme aracıdır. UDI taşıyıcıları, diğerlerine ilaveten, 1D (1-Boyutlu) / lineer barkod, 2D (2-Boyutlu) / Matriks barkod ve RFID şeklindedir. 2. Genel gereklilikler 2.1. UDI’nın iliştirilmesi ilave bir gereklilik olup bu Yönetmeliğin Ek I’inde belirtilen diğer işaretleme ve etiketleme gerekliliklerinin yerine geçmez. 2.2. İmalatçılar, cihazları için benzersiz UDI’ler tahsis eder ve bunları muhafaza eder. 2.3. Cihazın veya ambalajının üzerine sadece imalatçı UDI yerleştirebilir. 2.4. Sadece Komisyon tarafından atanan tahsis kuruluşlarınca sağlanan kodlama standartları kullanılabilir. 3. UDI 3.1. Bir UDI, cihazın kendisine veya ambalajına tahsis edilir. Ambalajın üst seviyelerinin kendilerine ait UDI’ları bulunur. 3.2. Nakliyat konteynırları, bu Kısmın 3.1. numaralı maddesindeki gereklilikten muaf tutulur. Örnek olarak, bir lojistik biriminde UDI’ya gerek yoktur; bir sağlık profesyonelinin münferit cihazların UDI’sını veya model numarasını kullanarak birden çok cihaz sipariş etmesi ve imalatçının bu cihazları nakliyat amacıyla ya da ayrı ayrı ambalajlanmış cihazları korumak için bir konteynıra koyması durumunda, konteynır (lojistik birim) UDI gerekliliklerine tabi değildir. 3.3. UDI, iki bölümden oluşur: UDI-DI ve UDI-PI. 3.4. UDI-DI, cihaz ambalajının her bir seviyesi için özgün olur. 3.5. Etiket üzerinde lot numarası, seri numarası, yazılım kimliği veya son geçerlilik tarihi yer alıyorsa bu, UDI-PI’ın bir parçası olur. Etiket üzerinde ayrıca bir imalat tarihi bulunuyorsa, UDI-PI’ya dâhil edilmesine gerek yoktur. Etiket üzerinde sadece imalat tarihi bulunuyorsa bu UDI-PI olarak kullanılır. 3.6. Bir cihaz olduğu kabul edilen ve kendi başına piyasada satılan her bir bileşene, kendi UDI’sı ile işaretlenmiş olan konfigüre edilebilir bir cihazın bileşenleri olmadıkça, farklı bir UDI tahsis edilir. 3.7. 22 nci maddede atıfta bulunulduğu şekilde sistemler ve işlem paketlerine kendi UDI’ları tahsis edilir ve bunlar kendi UDI’larını taşır. 3.8. İmalatçı, bir cihaza UDI tahsis ederken ilgili kodlama standardını izler. 3.9. Cihazın yanlış tanımlanmasına ve/veya izlenebilirliğinde belirsizliğe yol açabilecek bir değişiklikte, özellikle de aşağıdaki UDI veri tabanı veri ögelerinden herhangi birindeki durum değişikliklerinde yeni bir UDI-DI gerekir: a) Cihazın adı veya ticari adı, b) Cihaz versiyonu veya modeli, c) Tek kullanımlık olarak etiketlenmiş, ç) Steril ambalajlanmış, d) Kullanımdan önce sterilizasyon ihtiyacı, e) Ambalaj içindeki cihaz miktarı, f) Kritik uyarılar veya kontrendikasyonlar: örneğin, lateks veya DEHP içermesi. 3.10. Kendi etiketleriyle cihazları yeniden ambalajlayan ve/veya yeniden etiketleyen imalatçılar; orijinal cihaz imalatçısının UDI’sı ile ilgili kaydı tutar. 4. UDI taşıyıcısı 4.1. UDI taşıyıcısı (UDI’nın AIDC ve HRI gösterimi), etiket üzerine veya cihazın kendisi üzerine ve cihaz ambalajının daha üst seviyelerinin tamamının üzerine yerleştirilir. Üst seviyeler, nakliyat konteynırlarını kapsamaz. 4.2. Kullanım birimi ambalajı üzerinde bariz boşluk kısıtlaması olması durumunda, UDI taşıyıcısı, bir üst ambalaja yerleştirilebilir. 4.3. Sınıf I ve IIa tek kullanımlık cihazlar ayrı ayrı ambalajlanmış ve etiketlenmiş ise UDI taşıyıcısının bu ambalaj üzerinde bulunması gerekmez; fakat ambalajın üst seviyesinde örneğin, ayrı ayrı ambalajlanmış birkaç cihaz içeren bir karton kutu üzerinde UDI taşıyıcısı bulunur. Ancak sağlık profesyonelinin cihaz ambalajının üst seviyesini göremeyeceği bir durumda örneğin evde sağlık bakımı hizmetleri gibi durumlarda, UDI, her bir cihazın ambalajı üzerine yerleştirilir. 4.4. Yalnızca perakende satış noktasına yönelik cihazlar için satış noktası ambalajı üzerinde AIDC’deki UDI-PI’ların bulunması gerekmez. 4.5. UDI taşıyıcısından farklı bir AIDC taşıyıcısı ürün etiketlemesinin bir parçası olduğunda, UDI taşıyıcısı kolaylıkla belirlenebilir olur. 4.6. Lineer barkodlar kullanılıyorsa UDI-DI ve UDI-PI, iki veya daha fazla barkod üzerinde birleştirilmiş olabilir ya da ayrı ayrı bulunabilir. Lineer barkodun tüm bölümleri ve ögeleri, ayırt edilebilir ve belirlenebilir olur. 4.7. Etiket üzerinde AIDC ve HRI’nın birlikte kullanımını sınırlayan önemli kısıtlamalar varsa sadece AIDC formatının etiket üzerinde görünmesi gerekir. Evde bakım cihazları gibi, sağlık tesisleri dışında kullanılması amaçlanan cihazlar için HRI; AIDC için hiçbir boşluk kalmayacak olsa bile etiket üzerinde mutlaka bulunur. 4.8. HRI formatı, UDI kodu tahsis kuruluşunun kurallarını izler. 4.9 İmalatçı RFID teknolojisi kullanıyorsa, kod tahsis kuruluşları tarafından sağlanan standart doğrultusunda bir lineer veya 2-boyutlu barkod da etiket üzerinde bulunur. 4.10. Tekrar kullanılabilir cihazlar; cihazın kendisi üzerinde bir UDI taşıyıcısı taşır. Hastadan hastaya kullanımlar arasında temizlenmesi, dezenfekte edilmesi, steril edilmesi veya yenileştirilmesi gereken tekrar kullanılabilir cihazlara yönelik UDI taşıyıcısı; cihazın amaçlanan kullanım ömrü boyunca, sonraki kullanıma hazır hale getirilmesi için gerçekleştirilen her işlem sonrasında kalıcı ve okunabilir olur. Bu gereklilik, aşağıdaki durumlarda cihazlara uygulanmaz: a) Doğrudan işaretleme tiplerinin cihazın güvenliliği veya performansı ile çatıştığı durumda, b) Teknolojik olarak yapılabilir olmadığı için cihazın doğrudan işaretlenemediği durumda. 4.11. UDI taşıyıcısı, cihazın normal kullanımı sırasında ve öngörülen kullanım ömrü boyunca okunabilir olur. 4.12. Cihazın ambalajı UDI taşıyıcısının kolaylıkla okunabilmesine ya da AIDC söz konusu olduğunda taranabilmesine engel oluşturmuyor ise ayrıca ambalaj üzerine UDI taşıyıcısının yerleştirilmesi gerekmez. 4.13. İlk kullanımdan önce birleştirilmesi gereken birçok parçadan oluşan tek bir bitmiş cihaz söz konusu olduğunda, cihazın sadece bir parçasına UDI taşıyıcısı yerleştirmek yeterli olur. 4.14. UDI taşıyıcısı, cihazın normal çalışması veya depolanması süresince AIDC’ye erişilebilecek bir şekilde yerleştirilir. 4.15. Hem UDI-DI hem de UDI-PI içeren barkod taşıyıcıları, cihazın çalışması için gerekli verileri veya diğer verileri de içerebilir. 5. UDI veri tabanının genel ilkeleri 5.1. UDI veri tabanı, bu Ekin Kısım B’sinde atıfta bulunulan temel UDI veri tabanı veri ögelerinin tamamının kullanımını destekler. 5.2. İmalatçılar, UDI veri tabanındaki tanımlayıcı bilgilerin ve diğer cihaz veri ögelerinin ilk girişinden ve güncellenmelerinden sorumludur. 5.3. Elde edilen verilerin validasyonu için uygun yöntemler/prosedürler uygulanır. 5.4. İmalatçılar, artık piyasada bulunmayan cihazlar hariç olmak üzere, piyasaya arz ettikleri cihazlarla ilgili verilerin tümünün doğruluğunu periyodik olarak teyit eder. 5.5. UDI veri tabanında cihaz UDI-DI’sının bulunması, cihazın bu Yönetmeliğe uygun olduğu anlamına gelmez. 5.6. Veri tabanı, cihazın bütün ambalaj seviyeleri arasında bağlantı kurmaya imkân verir. 5.7. Yeni UDI-DI’lara yönelik veriler, cihazın piyasaya arz edildiği anda mevcut olur. 5.8. İmalatçılar, yeni bir UDI-DI gerektirmeden bir ögede yapılan değişiklikten itibaren 30 gün içerisinde ilgili UDI veri tabanı kaydını günceller. 5.9. Veri girişi ve güncellemelerine yönelik uluslararası kabul görmüş standartlar, mümkün olan her durumda, UDI veri tabanı tarafından kullanılır. 5.10. Artık piyasada bulunmayan cihazlarla ilgili veriler, UDI veri tabanında muhafaza edilir. 6. Spesifik cihaz tiplerine yönelik kurallar 6.1. İmplante edilebilir cihazlar 6.1.1. İmplante edilebilir cihazlar; ambalajlarının en alt seviyesinde (birim ambalajları), bir UDI (UDI-DI + UDI-PI) ile tanımlanır ya da AIDC kullanılarak işaretlenir. 6.1.2. UDI-PI, asgari olarak aşağıdaki karakteristiklere sahip olur: (a) İmplante edilebilir aktif cihazlar için seri numarası, (b) İmplante edilebilir diğer cihazlar için seri numarası veya lot numarası. 6.1.3. İmplante edilebilir cihazın UDI’sı, implantasyondan önce tanımlanabilir olur. 6.2. Kullanımlar arasında temizlik, dezenfeksiyon, sterilizasyon veya yenileştirme gerektiren tekrar kullanılabilir cihazlar 6.2.1. Bu tür cihazların UDI’sı cihaz üzerine yerleştirilir ve cihazı sonraki kullanıma hazır hale getirmeye yönelik her işlemden sonra okunabilir olur. 6.2.2. Lot veya seri numarası gibi UDI-PI karakteristikleri imalatçı tarafından belirlenir. 6.3. 22 nci maddede atıfta bulunulduğu şekilde sistemler ve işlem paketleri 6.3.1. 22 nci maddede atıfta bulunulan gerçek veya tüzel kişi; sistem veya işlem paketini, hem UDI-DI hem de UDI-PI içeren bir UDI ile tanımlamaktan sorumlu olur. 6.3.2. Sistem veya işlem paketlerinin cihaz bileşenleri ambalajlarının üzerinde veya cihazın kendi üzerinde bir UDI taşıyıcısı taşır. İstisnalar: a) Kullanımları, bu cihazları kullanması amaçlanan kişiler tarafından genel olarak bilinen, bir sistem veya işlem paketi içerisinde yer alan ve sistem veya işlem paketi kapsamı dışında tek başına kullanımı amaçlanmayan, her bir tek kullanımlık cihazın kendi başına UDI taşıyıcısı taşıması gerekmez; b) İlgili ambalaj seviyesinde bir UDI taşıyıcısı taşımaktan muaf tutulan cihazların, bir sistem veya işlem paketi içerisine dâhil edildiklerinde, bir UDI taşıyıcısı taşıması gerekmez. 6.3.3 UDI taşıyıcısının sistemlerin veya işlem paketlerinin üzerine yerleştirilmesi a) Sistem veya işlem paketinin UDI taşıyıcısı, genel bir kural olarak, ambalajın dışına iliştirilir. b) UDI taşıyıcısı; sistem veya işlem paketi ambalajının dışına iliştirilmiş olması ya da şeffaf ambalajın içerisinde bulunması durumlarında, okunabilir ya da AIDC söz konusu olduğunda taranabilir olur. 6.4. Konfigüre edilebilir cihazlar 6.4.1. Bir UDI, konfigüre edilebilir cihazın bütününe atanır ve konfigüre edilebilir cihaz UDI’sı olarak adlandırılır. 6.4.2. Konfigüre edilebilir cihaz UDI-DI’sı, grup içindeki her konfigürasyon başına değil, konfigürasyon gruplarına atanır. Bir konfigürasyon grubu, teknik dokümantasyonda açıklandığı şekilde belirli bir cihaza yönelik olası konfigürasyonların bütünü olarak tanımlanır. 6.4.3. Bir konfigüre edilebilir cihaz UDI-PI’sı, her bir münferit konfigüre edilebilir cihaza atanır. 6.4.4. Konfigüre edilebilir cihaz UDI’sının taşıyıcısı, sistemin kullanım ömrü boyunca değiştirilmesi en olasılık dışı olan bileşenin üzerine yerleştirilir ve konfigüre edilebilir cihaz UDI’sı olarak tanımlanır. 6.4.5. Bir cihaz olduğu kabul edilen ve kendi başına ticari olarak bulundurulan her bir bileşene farklı bir UDI atanır. 6.5. Cihaz yazılımı 6.5.1. UDI tahsis kriterleri UDI, yazılımın sistem seviyesinde atanır. Sadece kendi başına ticari olarak bulundurulan yazılım ve kendi başına bir cihaz olan yazılım bu gerekliliğe tabi olur. Yazılım kimliği, imalat kontrol mekanizması olarak kabul edilir ve UDI-PI’da gösterilir. 6.5.2. Aşağıdakileri değiştiren bir modifikasyon olduğunda, yeni bir UDI-DI gerekir: a) Orijinal performansı, b) Yazılımın güvenliğini veya kullanım amacını, c) Verilerin yorumlanmasını. Bu tür modifikasyonlar; yeni veya modifiye edilmiş algoritmalar, veri tabanı yapıları, işletim platformu, mimari veya yeni kullanıcı arayüzleri ya da birlikte çalışabilirliğe yönelik yeni kanallar içerir. 6.5.3. Minör yazılım revizyonları, yeni bir UDI-PI gerektirip yeni bir UDI-DI gerektirmez. Minör yazılım revizyonları; genel olarak hata (bug) düzeltmeleri, güvenlik amaçlı olmayan kullanılabilirlik iyileştirmeleri, güvenlik yamaları veya işletim verimliliği ile ilişkili olur. Minör yazılım revizyonları, imalatçıya özel tanımlama formu yoluyla tanımlanır. 6.5.4. Yazılıma yönelik UDI yerleştirme kriterleri a) Yazılımın, CD veya DVD gibi fiziksel bir ortamda verilmesi durumunda, her ambalaj seviyesi, UDI’nın tamamının HRI gösterimini ve AIDC gösterimini taşır. Yazılımı barındıran fiziksel ortama ve ambalajına uygulanan UDI, yazılıma sistem seviyesinde atanan UDI ile özdeş olur, b) UDI; kullanıcı için rahatlıkla ulaşılabilir bir ekran üzerinde, “hakkında” dosyası gibi, kolaylıkla okunabilir bir düz metin formatında sağlanır ya da başlangıç ekranına dâhil edilir, c) Görüntü dönüştürmeye yönelik ara katman (aracı) yazılımı gibi kullanıcı arayüzü olmayan bir yazılım, bir uygulama programlama arayüzü (API) vasıtasıyla UDI’yı iletme kabiliyetine sahip olur, ç) Yazılımın elektronik ekranlarında, yalnızca UDI’nın insan tarafından okunabilir kısmı gereklidir. “Hakkında” menüsü, açılır pencere vb. elektronik ekranlarda, AIDC kullanan UDI işaretine gerek yoktur, d) Yazılıma yönelik UDI’nın insan tarafından okunabilir formatı; UDI’yı tanımlamada ve UDI oluşturmak üzere kullanılan standardı belirlemede kullanıcıya yardımcı olması için kod tahsis kuruluşları tarafından kullanılan standarda yönelik Uygulama Tanımlayıcılarını (AI) içerir. 6.6. İleri düzeyde tekilleştirilen
cihazlar 6.6.1. Kontakt lensler 6.6.1.1. Standart kontakt lensler Asgari
olarak taban eğrisi ve çap dâhil olmak üzere, kontakt lens tasarım
parametreleri bakımından aynı kombinasyona sahip standart
kontakt lenslere bir UDI-DI tahsis edilir (Ana UDI-DI). 3.9
numaralı maddede belirtilen gerekliliğe ilave olarak, birinci
paragrafta atıfta bulunulan tasarım parametrelerinin
kombinasyonunda bir değişiklik olduğunda yeni bir Ana UDI-DI
gerekir. 6.6.1.2.Sipariş üzerine imal edilen kontakt lensler Asgari
olarak taban eğrisi ve çap dâhil olmak üzere kontakt lens tasarım
parametrelerinin aynı kombinasyonuna sahip ısmarlama imal edilen
kontakt lenslere bir UDI-DI tahsis edilir (Ana UDI-DI). 3.9 numaralı maddede belirtilen gerekliliğe ilave olarak, birinci paragrafta atıfta bulunulan tasarım parametrelerinin kombinasyonunda bir değişiklik olduğunda yeni bir Ana UDI-DI gerekir. (9/11/2025 tarihinde yürürlüğe girer)
____________________________________________
Ek VII ONAYLANMIŞ KURULUŞLAR TARAFINDAN KARŞILANACAK GEREKLİLİKLER 1. Organizasyonel ve genel gereklilikler 1.1. Hukuki statü ve organizasyonel yapı 1.1.1. Her onaylanmış kuruluş, ilgili mevzuat doğrultusunda kurulur ve onaylanmış kuruluşun tüzel kişiliği ve statüsü tamamıyla dokümante edilir. Bu tür dokümantasyon, sahiplik ve onaylanmış kuruluş üzerinde kontrolü bulunan gerçek veya tüzel kişiler hakkında bilgiler içerir. 1.1.2. Onaylanmış kuruluş, daha büyük bir kuruluşun parçası olan bir tüzel kişilik ise organizasyonel yapısı ve yönetimi ile birlikte bu kuruluşun faaliyetleri ve onaylanmış kuruluş ile ilişkisi açıkça dokümante edilir. Bu gibi durumlarda, bu Ekin 1.2 numaralı maddesi ile ilgili gereklilikler, hem onaylanmış kuruluş için hem de ait olduğu kuruluş için uygulanabilir olur. 1.1.3. Bir onaylanmış kuruluş, yurt dışında yerleşik olan tüzel kişiliklere kısmen veya tamamen sahipse ya da başka bir tüzel kişiliğin sahipliğindeyse, onaylanmış kuruluşla olan hukuki ve işlevsel ilişkileri ile birlikte bu tüzel kişiliklerin faaliyetleri ve sorumlulukları açıkça tanımlanır ve dokümante edilir. Bu Yönetmelik kapsamında uygunluk değerlendirme faaliyetleri yürüten söz konusu tüzel kişiliklerin personeli, bu Yönetmeliğin uygulanabilir gerekliliklerine tabi olur. 1.1.4. Onaylanmış kuruluşun organizasyonel yapısı, sorumlulukların tahsisi, raporlama yolları ve kuruluşun işleyişi; onaylanmış kuruluşun performansının ve yürüttüğü uygunluk değerlendirme faaliyetlerinin sonuçlarının güvenilir olmasını sağlayacak şekilde olur. 1.1.5. Onaylanmış kuruluş; organizasyonel yapısı ile üst yönetimin ve onaylanmış kuruluşun performansı ve uygunluk değerlendirme faaliyetlerinin sonuçları üzerinde bir etkiye sahip olabilecek diğer personelin görevlerini, sorumluluklarını ve yetkilerini açıkça dokümante eder. 1.1.6. Onaylanmış kuruluş, aşağıdakilerin her biri için bütün yetki ve sorumluluğa sahip olan üst yönetimdeki kişileri tanımlar: - Uygunluk değerlendirme faaliyetlerine yönelik yeterli kaynakların temini, - Onaylanmış kuruluşun işleyişine yönelik prosedürlerin ve politikaların geliştirilmesi, - Onaylanmış kuruluşun prosedürlerinin, politikalarının ve kalite yönetim sistemlerinin uygulanmasının kontrolü, - Onaylanmış kuruluşun mali durumunun kontrolü, - Sözleşmeye dayalı anlaşmalar dâhil olmak üzere, onaylanmış kuruluş tarafından yapılan faaliyetler ve alınan kararlar, - Gerekli olduğu durumlarda, tanımlanmış faaliyetlerin gerçekleştirilmesi için personele ve/veya komitelere yetki devri, - Kurum ile iş birliği ve diğer yetkili otoriteler, Komisyon ve diğer onaylanmış kuruluşlar ile iletişime ilişkin yükümlülükler. 1.2. Bağımsızlık ve tarafsızlık 1.2.1. Onaylanmış kuruluş; yürüttüğü uygunluk değerlendirme faaliyetlerine ilişkin olarak cihazın imalatçısından bağımsız bir üçüncü taraf kuruluş olur. Onaylanmış kuruluş, imalatçının rakiplerinin yanı sıra cihazdan bir çıkarı olan diğer iktisadi işletmecilerden de bağımsız olur. Bu durum, onaylanmış kuruluşun rakip imalatçılar için uygunluk değerlendirme faaliyetleri yürütmesine engel olmaz. 1.2.2. Onaylanmış kuruluş, faaliyetlerinin bağımsızlığını, objektifliğini ve tarafsızlığını koruyacak şekilde organize edilir ve işletilir. Onaylanmış kuruluş; tarafsızlığı korumaya yönelik ve organizasyon, personel ve değerlendirme faaliyetleri genelinde tarafsızlık ilkelerini desteklemeye ve uygulamaya yönelik bir yapıyı ve prosedürleri dokümante eder ve uygular. Bu tür prosedürler; onaylanmış kuruluşla çalışmaya başlamadan önce cihazlar alanında danışmanlık hizmetlerinde yer almak da dâhil olmak üzere, bir çıkar çatışmasının ortaya çıkabileceği her bir durumun tanımlanmasını, araştırılmasını ve çözülmesini sağlar. Bu araştırma, çıktı ve çözümleri dokümante edilir. 1.2.3. Onaylanmış kuruluş, üst yönetimi ve uygunluk değerlendirme görevlerini yürütmekten sorumlu personel: a) Değerlendirdikleri cihazların tasarımcısı, imalatçısı, tedarikçisi, kurulumcusu, alıcısı, sahibi veya bakım yapanı olamaz; bununla birlikte söz konusu tarafların herhangi birinin yetkili temsilcisi de olamaz. Bu tür kısıtlamalar, onaylanmış kuruluşun çalışmaları ve uygunluk değerlendirmenin yürütülmesi için gerekli olan değerlendirilen cihazların satın alınmasını ve kullanılmasını ya da bu tür cihazların kişisel amaçlara yönelik kullanımını engellemez. b) Atandıkları cihazların; tasarımında, imalatında veya yapımında, pazarlanmasında, kurulumunda ve kullanımında ya da bakımında yer almaz ve bu faaliyetlerde bulunan tarafları temsil etmez, c) Atandıkları uygunluk değerlendirme faaliyetlerine ilişkin dürüstlükleriyle ve kararlarının bağımsızlığıyla çatışabilecek hiçbir faaliyette bulunmaz, ç) Bağımsızlıklarına, tarafsızlıklarına veya objektifliklerine olan güveni tehlikeye düşürebilecek hiçbir hizmeti teklif etmez veya sağlamaz. Özellikle değerlendirme kapsamındaki cihazların ya da süreçlerin tasarımı, yapımı, pazarlanması veya bakımı hakkında, imalatçıya, imalatçının yetkili temsilcisine, bir tedarikçiye veya bir ticari rakibe danışmanlık hizmetleri teklif etmez veya sağlamaz, d) (ç) bendinde atıfta bulunulduğu şekilde danışmanlık hizmetleri sağlayan hiçbir kuruluşla bağlantılı olmaz. Bu tür kısıtlamalar, müşteriye özel olmayan ve cihazların mevzuatıyla veya ilgili standartlarla ilişkili genel eğitim faaliyetlerini engellemez. 1.2.4. Bir onaylanmış kuruluşla çalışmaya başlamadan önce cihazlar alanında danışmanlık hizmetinde yer alma durumu, işe başlama anında tamamen dokümante edilir ve olası çıkar çatışmaları bu Ek uyarınca izlenir ve çözümlenir. Belirli bir müşteri tarafından önceden istihdam edilen ya da bir onaylanmış kuruluşla çalışmaya başlamadan önce bu belirli müşteriye cihazlar alanında danışmanlık hizmeti sağlayan personel, 3 yıllık bir süre için bu belirli müşteriye veya aynı gruba ait şirketlere yönelik uygunluk değerlendirme faaliyetlerine atanmaz. 1.2.5. Onaylanmış kuruluşların, üst yönetimlerinin ve değerlendirme personelinin tarafsızlığı garanti edilir. Bir onaylanmış kuruluşun üst yönetiminin ve değerlendirme personelinin yanı sıra değerlendirme faaliyetlerine dâhil olan yüklenicilerin ücret miktarı, değerlendirmelerin sonuçlarına bağlı olmaz. Onaylanmış kuruluşlar, üst yönetimlerinin çıkar çatışması beyanlarını kamuya açık hale getirir. 1.2.6. Bir onaylanmış kuruluş bir kamu kurumunun veya kuruluşunun sahipliğindeyse, bunlar ile onaylanmış kuruluş ve/veya Kurum arasında bağımsızlık ve çıkar çatışmasının olmaması sağlanır ve bu durum dokümante edilir. 1.2.7. Onaylanmış kuruluş; sahiplerinin faaliyetleri dâhil olmak üzere, şube/temsilciliklerinin veya yüklenicilerinin ya da herhangi bir ilişkili kuruluşun faaliyetlerinin, kendi uygunluk değerlendirme faaliyetlerinin bağımsızlığını, tarafsızlığını veya objektifliğini etkilememesini sağlar ve bunu dokümante eder. 1.2.8. Onaylanmış kuruluş; ücretlere ilişkin olarak Küçük ve Orta Büyüklükteki İşletmelerin Tanımı, Nitelikleri ve Sınıflandırılması Hakkında Yönetmelikte tanımlandığı şekilde küçük ve orta ölçekli işletmelerin çıkarlarını dikkate alarak tutarlı, adil ve makul hükümler ve şartlar dizisine uygun olarak çalışır. 1.2.9. Bu Ekin 1.2 numaralı maddesinde belirtilen gereklilikler, bir onaylanmış kuruluş ile uygunluk değerlendirmesi için başvuran bir imalatçı arasında teknik bilgi ve mevzuata ilişkin kılavuz paylaşımını hiçbir şekilde engellemez. 1.3. Gizlilik 1.3.1. Onaylanmış kuruluş; personelinin, komitelerinin, şube/temsilciliklerinin, yüklenicilerinin ve harici kuruluşların personelinin ya da herhangi bir ilişkili kuruluşun; ifşa etmenin kanunen gerekli olduğu durumlar haricinde, uygunluk değerlendirme faaliyetlerinin yürütülmesi süresince elde ettikleri bilgilerin gizliliğine riayet etmesini sağlayan dokümante edilmiş işleyen prosedürlere sahip olur. 1.3.2. Bir onaylanmış kuruluşun personeli; Kurum, AB üyesi ülkelerin onaylanmış kuruluşlardan sorumlu otoriteleri ve tıbbi cihaz yetkili otoriteleri veya Komisyon ile ilişkili görevleri hariç olmak üzere, bu Yönetmelik ya da kendisini etkileyen diğer mevzuat hükümleri kapsamında görevlerini yerine getirirken mesleki gizliliği gözetir. Mülkiyet hakları korunur. Onaylanmış kuruluş, bu Ekin 1.3 numaralı maddesinin gereklilikleriyle ilgili olarak dokümante edilmiş işleyen prosedürlere sahip olur. 1.4. Sorumluluk 1.4.1. İlgili mevzuat uyarınca sorumluluğun kamu tarafından üstlenildiği veya uygunluk değerlendirmesinden doğrudan kamunun sorumlu olduğu durumlar hariç olmak üzere, onaylanmış kuruluş uygunluk değerlendirme faaliyetlerine uygun şekilde sorumluluk sigortası yaptırır. 1.4.2. Sorumluluk sigortasının kapsamı ve tam mali değeri, onaylanmış kuruluşun faaliyetlerinin düzeyine ve coğrafik kapsamına karşılık gelir ve onaylanmış kuruluş tarafından belgelendirilen cihazların risk profiliyle orantılı olur. Sorumluluk sigortası, onaylanmış kuruluşun sertifikaları geri çekmek, kısıtlamak veya askıya almak zorunda kalabileceği durumları kapsar. 1.5. Mali gereklilikler Onaylanmış kuruluş; atanma kapsamı dâhilinde uygunluk değerlendirme faaliyetlerini ve ilgili ticari çalışmaları yürütmek için gerekli mali kaynaklara sahip olur. Onaylanmış kuruluş, ilgili olduğu yerde başlangıç aşamasındaki belirli durumları dikkate alarak mali kapasitesi ve uzun vadeli ekonomik yaşayabilirliği ile ilgili kanıtları dokümante eder ve sunar. 1.6. Koordinasyon faaliyetlerine katılım 1.6.1. Onaylanmış kuruluş; ilgili standardizasyon faaliyetlerine ve 49 uncu maddede atıfta bulunulan onaylanmış kuruluş koordinasyon grubu faaliyetlerine katılır ya da değerlendirme personelinin bu faaliyetler hakkında bilgilendirilmesini sağlar. Ayrıca değerlendirme ve karar alıcı personelinin bu Yönetmelik çerçevesinde kabul edilen ilgili bütün mevzuat, kılavuz ve iyi uygulama dokümanları konusunda bilgilendirilmesini sağlar. 1.6.2. Onaylanmış kuruluş, kılavuz ve iyi uygulama dokümanlarını dikkate alır. 2. Kalite yönetimi gereklilikleri 2.1. Onaylanmış kuruluş, uygunluk değerlendirme faaliyetlerinin niteliğine, alanına ve ölçeğine uygun olan ve bu Yönetmeliğin gerekliliklerinin sürekli olarak yerine getirilmesini destekleme ve gösterme kabiliyeti olan bir kalite yönetim sistemi oluşturur, dokümante eder, uygular, sürdürür ve işletir. 2.2. Onaylanmış kuruluşun kalite yönetim sistemi asgari olarak; - Faaliyetlerine yönelik politikalar ve hedefler dâhil olmak üzere, yönetim sistemi yapısına ve dokümantasyona, - Faaliyetlerin ve sorumlulukların personele tahsis edilmesine yönelik politikalara, - Onaylanmış kuruluşun personelinin ve üst yönetiminin görevlerine, sorumluluklarına ve rolüne uygun olarak; değerlendirme ve karar alma süreçlerine, - Uygunluk değerlendirme prosedürlerinin; planlanması, yürütülmesi, değerlendirilmesi ve gerektiğinde adaptasyonuna, - Dokümanların kontrolüne, - Kayıtların kontrolüne, - Yönetim gözden geçirmelerine, - İç denetimlere, - Düzeltici ve önleyici faaliyetlere, - Şikâyetler ve itirazlara, - Sürekli eğitime değinir. Dokümanların farklı dillerde kullanılması durumunda onaylanmış kuruluş, bunların aynı içeriğe sahip olduğunu garanti ve kontrol eder. 2.3. Onaylanmış kuruluşun üst yönetimi; bu Yönetmelik uyarınca uygunluk değerlendirme faaliyetlerinde yer alan şube/temsilcilikler ve yükleniciler dâhil olmak üzere onaylanmış kuruluşun bütününde, kalite yönetim sisteminin tam olarak anlaşıldığını, uygulandığını ve sürdürüldüğünü garanti eder. 2.4. Onaylanmış kuruluş, tüm personelinden onaylanmış kuruluş tarafından tanımlanan prosedürlere uymak üzere bir imza veya eşdeğeri yoluyla resmi olarak taahhütte bulunmasını talep eder. Bu taahhütname; gizlilik, ticari ve diğer çıkarlardan bağımsızlık ile müşterilerle mevcut ya da önceden var olmuş herhangi bir bağlantı ile ilgili hususları kapsar. Personelin; gizlilik, bağımsızlık ve tarafsızlık ilkelerine uyduğunu belirten yazılı beyanları vermesi zorunludur. 3. Kaynak gereklilikleri 3.1. Genel 3.1.1. Onaylanmış kuruluşlar; bu Yönetmelik kapsamında kendilerine verilen bütün görevleri, bu görevlerin onaylanmış kuruluşların kendileri tarafından yürütülmesine ya da onların adına ve onların sorumluluğu altında yürütülmesine bakılmaksızın, en yüksek düzeyde mesleki dürüstlük ve spesifik alanda gerekli yetkinlikle yerine getirme kabiliyetine sahip olur. Onaylanmış kuruluşlar özellikle; gerekli personele sahip olur ve atanmış oldukları uygunluk değerlendirme faaliyetlerinde zorunlu kılınan teknik, bilimsel ve idari görevleri uygun bir şekilde yerine getirmek için gerek duyulan bütün ekipmana, tesislere ve yetkinliğe sahip olur veya erişim sağlar. Bu gibi gereklilikler; onaylanmış kuruluşun, atanmış olduğu her uygunluk değerlendirme prosedürü ve her cihaz tipi için ilgili cihazlara ve benzer teknolojilere ilişkin bilgi ve deneyime sahip yeterli sayıda idari, teknik ve bilimsel personeli kalıcı olarak her daim bulundurmasını gerektirir. Bu tür personel; söz konusu onaylanmış kuruluşun, özellikle Ek I’de belirtilenler olmak üzere bu Yönetmeliğin gerekliliklerini göz önüne alarak cihazların tıbbi işlevselliğinin, klinik değerlendirmelerinin, performansının ve güvenliliğinin değerlendirilmesi de dâhil atanmış olduğu uygunluk değerlendirme görevlerini yerine getirebilmesini sağlamak için yeterli sayıda olur. Bir onaylanmış kuruluşun kümülatif yetkinlikleri, atandığı cihaz tiplerini değerlendirmesine imkân sağlayacak nitelikte olur. Onaylanmış kuruluş, dış uzman tarafından yürütülen değerlendirmeleri eleştirel bir yaklaşımla değerlendirmek üzere yeterli iç yetkinliğe sahip olur. Bir onaylanmış kuruluşun yükleniciye veremeyeceği görevler bu Ekin 4.1 numaralı maddesinde düzenlenmiştir. Onaylanmış kuruluşun cihazlara yönelik uygunluk değerlendirme faaliyetlerinin işleyişinin yönetiminde yer alan personel; değerlendirme ve doğrulama çalışmalarını yürüten ve gerçekleştiren personelin, onlara verilen görevleri yerine getirmeleri için yetkin olmalarını sağlamak amacıyla; değerlendirme ve doğrulama personelinin seçimine, yetkinliklerinin doğrulanmasına, yetkilendirilmelerine ve görevlerinin dağılımına, başlangıç ve sürekli eğitimlerinin organize edilmesine, görevlerinin atanmasına ve bu personelin izlenmesine yönelik bir sistem kurmak ve işletmek için uygun bilgiye sahip olur. Onaylanmış kuruluş, üst yönetimdeki en az bir kişiyi, cihazlarla ilgili tüm uygunluk değerlendirme faaliyetlerinin tamamından sorumlu olacak şekilde tanımlar. 3.1.2. Onaylanmış kuruluş; deneyim paylaşımına yönelik bir sistem ve sürekli bir eğitim öğretim programı uygulayarak uygunluk değerlendirme faaliyetlerine dâhil olan personelin yeterliliklerini ve uzmanlıklarını sürdürmesini sağlar. 3.1.3 Onaylanmış kuruluş; yükleniciler ve dış uzmanlar dâhil olmak üzere uygunluk değerlendirme faaliyetlerinde yer alan personelin, görevlerinin ve sorumluluklarının kapsamını ve sınırlarını ve yetkilendirme düzeyini açıkça dokümante eder ve söz konusu personeli bu doğrultuda bilgilendirir. 3.2. Personele ilişkin yeterlilik kriterleri 3.2.1. Onaylanmış kuruluş; bilgi, deneyim ve gerekli diğer yetkinlikler dâhil olmak üzere uygunluk değerlendirme faaliyetlerinde yer alan kişilerin seçimine ve yetkilendirilmesine yönelik yeterlilik kriterleri ve prosedürler ile gerekli başlangıç eğitimlerini ve sürekli eğitimleri oluşturur ve dokümante eder. Yeterlilik kriterleri: atama kapsamında yer alan; cihazlar, teknolojiler ve faaliyet alanına (örneğin biyouyumluluk, sterilizasyon, insan ve hayvan kaynaklı dokular ve hücreler, klinik değerlendirme vb.) ve bunlarla birlikte denetleme, ürün değerlendirme veya test etme, teknik dokümantasyon incelemesi ve karar alma gibi uygunluk değerlendirme süreci içerisindeki çeşitli fonksiyonlara değinir. 3.2.2. Bu Ekin 3.2.1 numaralı maddesinde atıfta bulunulan yeterlilik kriterleri; 42 nci maddenin üçüncü fıkrasında belirtilen bildirim için Kurum tarafından tanımlanan kapsam uyarınca bir onaylanmış kuruluşun atama kapsamına atıfta bulunur ve kapsam tanımının alt bölümlerindeki gerekli yeterliliğe yönelik uygun seviyede ayrıntı sağlar. Spesifik yeterlilik kriterleri, asgari olarak aşağıdakilerin değerlendirilmesine yönelik tanımlanır: - Klinik öncesi değerlendirme, - Klinik değerlendirme, - İnsan ve hayvan kaynaklı dokular ve hücreler, - Fonksiyonel güvenlilik, - Yazılım, - Ambalajlama, - Bir tıbbi ürünü bütünleşik bir parça olarak ihtiva eden cihazlar, - İnsan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar, - Sterilizasyon süreçlerinin farklı türleri. 3.2.3. Yeterlilik kriterlerini oluşturmaktan ve spesifik uygunluk değerlendirme faaliyetlerini gerçekleştirmek için diğer personeli yetkilendirmekten sorumlu personel; onaylanmış kuruluşun kendisi tarafından istihdam edilir ve dış uzman veya yüklenici personeli olamaz. Bu personel, aşağıdakilerin tamamında kanıtlanmış bilgi ve deneyime sahip olur: - Cihazlara ilişkin mevzuat ve ilgili kılavuz dokümanlar, - Bu Yönetmelikte belirtilen uygunluk değerlendirme prosedürleri, - Cihazların tasarımı ve imalatı ile birlikte cihaz teknolojileri hakkında kapsamlı bir bilgi altyapısı, - Onaylanmış kuruluşun kalite yönetim sistemi, ilgili prosedürler ve gerekli yeterlilik kriterleri, - Cihazlarla ilgili uygunluk değerlendirme faaliyetlerinde yer alan personele ilişkin eğitim, - Bir onaylanmış kuruluş bünyesinde bu Yönetmelik veya önceden uygulanan mevzuat kapsamındaki uygunluk değerlendirmelerinde yeterli deneyim. 3.2.4. Onaylanmış kuruluş, ilgili klinik uzmanlığa sahip personeli kalıcı olarak elinde bulundurur ve mümkün olduğu yerde bu tür personel onaylanmış kuruluşun kendisi tarafından istihdam edilir. Bu tür personel, aşağıdakileri yapmak için onaylanmış kuruluşun değerlendirme ve karar alma süreçlerine başından sonuna kadar dâhil edilir: - İmalatçı tarafından yürütülen klinik değerlendirmenin onaylanmış kuruluşça değerlendirilmesi için uzman girdisinin ne zaman gerektiğini tanımlamak ve nitelikli uzmanları uygun bir şekilde belirlemek, - Bu Yönetmeliğin, ortak spesifikasyonların, kılavuzların ve uyumlaştırılmış standartların ilgili gereklilikleri hakkında dış klinik uzmanları uygun bir şekilde eğitmek ve dış klinik uzmanların, değerlendirmelerinin ve verdikleri tavsiyelerin içeriğinin ve yansımalarının tamamen farkında olmalarını sağlamak, - Klinik değerlendirmenin ve ilişkili klinik araştırmaların içerdiği klinik verileri inceleyebilmek ve bilimsel olarak tartışabilmek ve imalatçı tarafından sunulan klinik değerlendirmenin değerlendirilmesinde dış klinik uzmanlara uygun bir şekilde rehberlik edebilmek, - Sunulan klinik değerlendirmeyi ve imalatçının klinik değerlendirmesi ile ilgili dış klinik uzmanların değerlendirme sonuçlarını bilimsel açıdan değerlendirebilmek ve gerektiğinde tartışabilmek, - Klinik uzmanlar tarafından yürütülen klinik değerlendirmenin değerlendirilmesinin tutarlılığını ve karşılaştırılabilirliğini belirleyebilmek, - İmalatçının klinik değerlendirmesi ile ilgili bir değerlendirme yapabilmek ve herhangi bir dış uzman tarafından sunulan görüş ile ilgili bir klinik yargıya varabilmek ve onaylanmış kuruluşun karar alıcısına bir tavsiyede bulunabilmek ve - İlgili uygunluk değerlendirme faaliyetlerinin uygun bir şekilde yürütülmüş olduğunu gösteren kayıtlar ve raporlar hazırlayabilmek. 3.2.5. Klinik değerlendirme, biyolojik güvenlik, sterilizasyon ve yazılım validasyonu gibi konular dâhil olmak üzere teknik dokümantasyon incelemeleri veya tip incelemesi gibi ürünle ilgili incelemeleri yürütmekten sorumlu personel (ürün inceleyici), aşağıdaki kanıtlanmış yeterliliklerin tümüne sahip olur: - İlgili alanlarda bir üniversite veya bir teknik yüksekokul programının ya da bunlara denk bir yeterliliğin başarılı bir şekilde tamamlanması, örneğin; tıp, diş hekimliği, eczacılık, mühendislik veya diğer ilgili bilimler, - Sağlık ürünleri alanında ya da ilişkili faaliyetlerde (örn. imalat, denetim veya araştırma gibi) 4 yıllık mesleki deneyim; bunun 2 yılı değerlendirilecek cihazın veya teknolojinin tasarımında, imalatında, test edilmesinde veya kullanımında olur ya da değerlendirilecek bilimsel konularla ilgili olur, - Ek I’de belirtilen genel güvenlilik ve performans gereklilikleri dâhil olmak üzere cihaz mevzuatı ile ilgili bilgi, - İlgili uyumlaştırılmış standartlar, ortak spesifikasyonlar ve kılavuz dokümanlar ile ilgili uygun bilgi ve deneyim, - Risk yönetimi, ilgili cihaz standartları ve kılavuz dokümanlar ile ilgili uygun bilgi ve deneyim, - Klinik değerlendirme ile ilgili uygun bilgi ve deneyim, - Değerlendirdikleri cihazlar ile ilgili uygun bilgi, - Özellikle sorumlu oldukları prosedürler ile ilgili hususlar olmak üzere Ek IX ila Ek XI’de belirtilen uygunluk değerlendirme prosedürleri hakkında uygun bilgi ve deneyim ve bu değerlendirmeleri yürütmek için yeterli yetkilendirme, - İlgili uygunluk değerlendirme faaliyetlerinin uygun bir şekilde yürütülmüş olduğunu gösteren kayıtları ve raporları hazırlama kabiliyeti. 3.2.6. İmalatçının kalite yönetim sisteminin denetimlerini yürütmekten sorumlu personel (saha denetçisi), aşağıdaki kanıtlanmış yeterliliklerin tümüne sahip olur: - İlgili alanlarda bir üniversite veya bir teknik yüksekokul programının ya da bunlara denk bir yeterliliğin başarılı bir şekilde tamamlanması, örneğin; tıp, diş hekimliği, eczacılık, mühendislik veya diğer ilgili bilimler, - Sağlık ürünleri alanında ya da ilişkili faaliyetlerde (örn. imalat, denetim veya araştırma gibi) 4 yıllık mesleki deneyim; bunun 2 yılı kalite yönetimi alanında olur, - İlgili uyumlaştırılmış standartlar, ortak spesifikasyonlar ve kılavuz dokümanlarla birlikte cihaz mevzuatı ile ilgili uygun bilgi, - Risk yönetimi, ilgili cihaz standartları ve kılavuz dokümanlar hakkında uygun bilgi ve deneyim, - Kalite yönetim sistemleri, ilgili standartlar ve kılavuz dokümanlar hakkında uygun bilgi, - Özellikle sorumlu oldukları prosedürler ile ilgili hususlar olmak üzere, Ek IX ila Ek XI’de belirtilen uygunluk değerlendirme prosedürleri hakkında uygun bilgi ve deneyim ve bu denetimleri yürütmek için yeterli yetkilendirme, - Kalite yönetim sistemlerini değerlendirmelerini mümkün kılan denetim teknikleri konusunda eğitim, - İlgili uygunluk değerlendirme faaliyetlerinin uygun bir şekilde yürütülmüş olduğunu gösteren kayıtları ve raporları hazırlama kabiliyeti. 3.2.7. Belgelendirme konusunda nihai incelemeler yapmak ve karar almak için tüm sorumluluğa sahip olan personel, onaylanmış kuruluşun kendisi tarafından istihdam edilir ve dış uzman veya yüklenici personeli olamaz. Bu personel, bir bütün olarak aşağıdakilerin tümü ile ilgili kanıtlanmış bilgiye ve kapsamlı deneyime sahip olur: - Cihaz mevzuatı ve ilgili kılavuz dokümanlar, - Bu Yönetmelik ile ilgili cihaz uygunluk değerlendirmeleri, - Cihaz uygunluk değerlendirmesi ile ilgili yeterliliklerin, deneyimin ve uzmanlığın türleri, - Belgelendirme için incelenen cihazların uygunluk değerlendirmesi ile ilgili yeterli deneyim dâhil olmak üzere cihaz teknolojileri, cihaz endüstrisi ve cihazların tasarımı ve imalatı hakkında kapsamlı bir bilgi altyapısı, - Onaylanmış kuruluşun kalite yönetim sistemi, ilgili prosedürler ve sisteme dâhil olan personel için gerekli yeterlilikler, - Uygunluk değerlendirme faaliyetlerinin uygun bir şekilde yürütülmüş olduğunu gösteren kayıtları ve raporları hazırlama kabiliyeti. 3.3. Personelin yeterliliği, eğitimi ve yetkilendirilmesi ile ilgili dokümantasyon 3.3.1. Onaylanmış kuruluş, uygunluk değerlendirme faaliyetlerine dâhil olan her bir personelin yeterliliğini ve bu Ekin 3.2 numaralı maddesinde atıfta bulunulan yeterlilik kriterlerinin yerine getirilmesini tam olarak dokümante etmek için işleyen bir prosedüre sahip olur. Bu Ekin 3.2 numaralı maddesinde belirtilen yeterlilik kriterlerinin yerine getirilmesinin tam olarak gösterilemediği istisnai durumlarda, onaylanmış kuruluş, spesifik uygunluk değerlendirme faaliyetlerini yürütmek üzere ilgili personelin yetkilendirilmesini Kuruma gerekçelendirir. 3.3.2. Bu Ekin 3.2.3 ila 3.2.7 numaralı maddelerinde atıfta bulunulan personelin tümü için, onaylanmış kuruluş aşağıdakileri oluşturur ve güncel tutar: - Uygunluk değerlendirme faaliyetleriyle ilgili olarak personelin yetkilerini ve sorumluluklarını detaylandıran bir matris, - Yetkilendirildikleri uygunluk değerlendirme faaliyeti için gerekli bilgi ve deneyimi kanıtlayan kayıtlar. Bu kayıtlar; her bir değerlendirme personeline ilişkin sorumlulukların kapsamını belirlemeye yönelik bir gerekçe ve her birinin yürüttüğü uygunluk değerlendirme faaliyetlerinin kayıtlarını içerir. 3.4. Yükleniciler ve dış uzmanlar 3.4.1. Onaylanmış kuruluşlar, bu Ekin 3.2 numaralı maddesine halel gelmeksizin, bir uygunluk değerlendirme faaliyetinin açıkça tanımlanmış, belirli ve tamamlayıcı bölümlerini yükleniciye verebilir. Kalite yönetim sistemlerinin denetimi veya ürünle ilgili incelemeler bir bütün halinde yükleniciye verilemez; ancak bu faaliyetlerin bölümleri, yükleniciler ve onaylanmış kuruluş adına çalışan dış denetçiler ve uzmanlar tarafından yürütülebilir. Spesifik görevlerini yerine getirmeleri için yüklenicilerin ve uzmanların yetkinlikleri hakkında uygun kanıtlar üretebilmeye, bir yüklenicinin değerlendirmesine dayalı olarak bir karar almaya ve onaylanmış kuruluş adına yükleniciler ve uzmanlar tarafından yürütülen işe yönelik tüm sorumluluk söz konusu onaylanmış kuruluşta kalmaya devam eder. Aşağıdaki faaliyetler, onaylanmış kuruluşlar tarafından yükleniciye verilemez: - Dış uzmanların yeterliliklerinin incelenmesi ve performanslarının izlenmesi, - Söz konusu yükleniciliğin denetim veya belgelendirme kuruluşlarına verilmesi durumunda, denetim ve belgelendirme faaliyetleri, - Spesifik uygunluk değerlendirme faaliyetleri için dış uzmanlara işin tahsis edilmesi, - Nihai inceleme ve karar alma fonksiyonları. 3.4.2. Bir onaylanmış kuruluşun belirli uygunluk değerlendirme faaliyetlerini yüklenici olarak bir kuruluşa veya bir kişiye vermesi durumunda, onaylanmış kuruluş, yükleniciliğin hangi şartlar altında oluşabileceğini açıklayan bir politikaya sahip olur ve aşağıdakileri sağlar: - Yüklenicinin bu Ekin ilgili gerekliliklerini karşılamasını, - Yüklenicilerin ve dış uzmanların, işi yüklenici olarak başka kuruluşlara veya personele devretmemesini, - Uygunluk değerlendirmesi için başvuru yapan gerçek veya tüzel kişinin, birinci ve ikinci alt bentlerde atıfta bulunulan gereklilikler hakkında bilgilendirilmesini. Herhangi bir yüklenicilik ve dış personel danışmanlığı uygun bir şekilde dokümante edilir, hiçbir aracı içermez ve diğer hususların yanı sıra gizlilik ve çıkar çatışmasını da kapsayan yazılı bir anlaşmaya tabi olur. Söz konusu onaylanmış kuruluş, yükleniciler tarafından gerçekleştirilen görevlerin bütün sorumluluğunu üstlenir. 3.4.3. Yüklenicilerin veya dış uzmanların özellikle yeni, invaziv ve implante edilebilir cihazlar veya teknolojilerle ilgili bir uygunluk değerlendirmesi kapsamında kullanılması durumunda, söz konusu onaylanmış kuruluş, atandığı her ürün alanında; uygunluk değerlendirmesinin bütününü yönetmek, uzman görüşlerinin uygunluğunu ve geçerliliğini doğrulamak ve belgelendirme konusunda karar almak için yeterli iç yetkinliğe sahip olur. 3.5. Yetkinliklerin izlenmesi, eğitim ve deneyim paylaşımı 3.5.1. Onaylanmış kuruluş; uygunluk değerlendirme faaliyetlerine dâhil olan bütün iç ve dış personelin ve yüklenicilerin yetkinliklerinin, uygunluk değerlendirme faaliyetlerinin ve performansının ilk değerlendirmesine ve sürekli izlenmesine yönelik prosedürler oluşturur. 3.5.2. Onaylanmış kuruluşlar, düzenli aralıklarla, personelinin yetkinliklerini gözden geçirir, eğitim ihtiyaçlarını belirler ve her bir personelin gereken yeterlilik ve bilgi seviyesini sürdürmesi için bir eğitim planı hazırlar. Bu gözden geçirme, asgari olarak aşağıdaki hususları doğrular: - Personelin, cihazlara ilişkin mevzuattan, ilgili uyumlaştırılmış standartlardan, ortak spesifikasyonlardan, kılavuz dokümanlardan ve bu Ekin 1.6 numaralı maddesinde atıfta bulunulan koordinasyon faaliyetlerinin sonuçlarından haberdar olması, - Personelin, bu Ekin 3.1.2 numaralı maddesinde atıfta bulunulan iç deneyim paylaşımına ve sürekli eğitim öğretim programına katılması. 4. Süreç gereklilikleri 4.1. Genel Onaylanmış kuruluş; başvuru öncesi faaliyetlerinden karar alma ile gözetim ve denetime kadar her bir aşamayı kapsayan ve gerektiğinde cihazların kendi özgünlüklerini dikkate alarak atandığı her bir uygunluk değerlendirme faaliyetini yürütmeye yönelik dokümante edilmiş işleyen süreçlere ve yeterli bir şekilde detaylandırılmış prosedürlere sahip olur. Bu Ekin 4.3, 4.4, 4.7 ve 4.8 numaralı maddelerinde belirtilen gereklilikler, onaylanmış kuruluşların iç faaliyetlerinin parçası olarak yerine getirilir ve yükleniciye verilemez. 4.2. Onaylanmış kuruluş fiyat tarifesi ve başvuru öncesi faaliyetleri Onaylanmış kuruluş: a) İmalatçıların, kendisinden sertifika alabileceği başvuru prosedürünün açıklamasını kamuya açık bir şekilde yayımlar. Bu açıklama, dokümantasyon sunumu ve ilgili yazışmalar için hangi dillerin kabul edilebilir olduğunu belirtir, b) Belirli uygunluk değerlendirme faaliyetleri için uygulanan ücretlere ve cihazlara yönelik onaylanmış kuruluşun değerlendirme faaliyetleri ile ilgili diğer mali şartlara ilişkin dokümante edilmiş prosedürlere ve bu hususlar hakkında dokümante edilmiş detaylara sahip olur, c) Uygunluk değerlendirme hizmetlerinin reklamıyla ilgili dokümante edilmiş prosedürlere sahip olur. Bu prosedürler; reklam veya tanıtım faaliyetlerinin, kendi uygunluk değerlendirmelerinin imalatçılara daha erken piyasa erişimi sunacağı ya da diğer onaylanmış kuruluşlara nazaran daha hızlı, daha kolay veya daha az zorlayıcı olacağı çıkarımını hiçbir şekilde ima etmemesini ya da böyle bir çıkarıma yol açmamasını sağlar, ç) Spesifik bir uygunluk değerlendirmeyle ilgili olarak imalatçıya bir fiyat vermeden önce, ürünün bu Yönetmeliğin kapsamında olduğuna ve sınıflandırmasına dair ön doğrulama dâhil olmak üzere başvuru öncesi bilgilerin incelenmesini gerektiren dokümante edilmiş prosedürlere sahip olur, d) Bu Yönetmelik kapsamındaki uygunluk değerlendirme faaliyetlerine ilişkin bütün sözleşmelerin diğer kuruluşlarla değil doğrudan imalatçı ile onaylanmış kuruluş arasında akdedilmesini sağlar. 4.3. Başvurunun incelenmesi ve sözleşme Onaylanmış kuruluş; Ek IX ila Ek XI’de atıfta bulunulduğu şekilde ilgili uygunluk değerlendirmesinin gerektirdiği tüm bilgileri ve imalatçının beyanlarını içeren, imalatçı veya yetkili temsilci tarafından imzalanan resmi bir başvuru talep eder. Bir onaylanmış kuruluş ile bir imalatçı arasındaki sözleşme, her iki tarafça imzalanan yazılı bir anlaşma biçiminde olur. Sözleşme, onaylanmış kuruluş tarafından muhafaza edilir. Bu sözleşme, açık hüküm ve şartlar içerir ve imalatçının vijilans raporları hakkında onaylanmış kuruluşu bilgilendirme yükümlülüğü, onaylanmış kuruluşun düzenlenen sertifikaları askıya alma, kısıtlama veya geri çekme hakkı ve onaylanmış kuruluşun bilgilendirme yükümlülüklerini yerine getirme görevi dâhil olmak üzere, onaylanmış kuruluşun bu Yönetmelik kapsamında gerektiği şekilde hareket etmesine olanak sağlayan yükümlülükleri içerir. Onaylanmış kuruluş, başvuruları incelemek için aşağıdakileri ele alan dokümante edilmiş prosedürlere sahip olur: a) Onay istenen ilgili Ekte atıfta bulunulduğu şekilde, ilgili uygunluk değerlendirme prosedürünün gerekliliklerine ilişkin olarak başvuruların eksiksiz olması, b) Başvuruların kapsadığı ürünlerin cihaz olarak değerlendirilip değerlendirilemeyeceğinin ve bunların sınıflandırmalarının doğrulanması, c) Başvuru sahibi tarafından tercih edilen uygunluk değerlendirme prosedürlerinin bu Yönetmelik kapsamında söz konusu cihaza uygulanabilir olup olmaması, ç) Onaylanmış kuruluşun, atandığı kapsamda başvuruyu değerlendirme kabiliyeti, d) Yeterli ve uygun kaynakların mevcudiyeti. Bir başvuru ile ilgili her bir incelemenin çıktısı dokümante edilir. Başvuruların reddedilmesi veya geri çekilmesi, 57 nci maddede atıfta bulunulan elektronik sisteme girilir ve diğer onaylanmış kuruluşlar için erişilebilir olur. 4.4. Kaynakların tahsisi Onaylanmış kuruluş, tüm uygunluk değerlendirme faaliyetlerinin; uygunluk değerlendirmeye konu olan cihazlar, sistemler, süreçler ve ilgili dokümantasyonun değerlendirilmesinde yeterince deneyimli olan ve uygun bir şekilde yetkilendirilmiş nitelikli personel tarafından yürütülmesini sağlamak üzere dokümante edilmiş prosedürlere sahip olur. Onaylanmış kuruluş, her bir başvuru için gerekli kaynakları belirler ve bu başvurunun değerlendirmesinin ilgili prosedürler uyarınca yürütülmesini sağlamaktan ve değerlendirme görevlerinin her biri için kullanılan personel dâhil olmak üzere uygun kaynakları sağlamaktan sorumlu bir kişi tanımlar. Uygunluk değerlendirmesinin bir bölümü olarak yürütülmesi gereken görevlerin dağılımı ve bu dağılımda sonradan yapılan değişiklikler dokümante edilir. 4.5. Uygunluk değerlendirme faaliyetleri 4.5.1. Genel Onaylanmış kuruluş ve personeli, uygunluk değerlendirme faaliyetlerini yüksek düzeyde mesleki dürüstlükle ve spesifik alanlarda gerekli teknik ve bilimsel yetkinlikle yürütür. Onaylanmış kuruluş, Ek IX ila Ek XI’de belirtilen ilgili gereklilikleri ve özellikle aşağıdaki gerekliliklerin tümünü dikkate alarak atandığı uygunluk değerlendirme faaliyetlerini etkili bir şekilde yürütmek için yeterli uzmanlığa, tesislere ve dokümante edilmiş prosedürlere sahip olur: - Her bir projenin yürütülmesini uygun şekilde planlamak, - Değerlendirme ekiplerinin yapısını ilgili teknolojiye ilişkin yeterli deneyim olacak şekilde sağlamak ve objektiflik ve bağımsızlığı sürekli olacak şekilde temin etmek ve değerlendirme ekibi üyelerinin uygun aralıklarla rotasyonunu yapmak, - Uygunluk değerlendirme faaliyetlerinin tamamlanmasına yönelik süre sınırları saptamak için gerekçe belirtmek, - İmalatçının teknik dokümantasyonunu ve Ek I’de belirtilen gereklilikleri karşılamak için benimsenen çözümleri değerlendirmek, - Klinik öncesi hususların değerlendirilmesiyle ilgili imalatçının prosedürlerini ve dokümantasyonunu incelemek, - Klinik değerlendirme ile ilgili imalatçının prosedürlerini ve dokümantasyonu incelemek, - İmalatçının risk yönetimi süreci ve bu sürecin değerlendirilmesi ile klinik öncesi ve klinik değerlendirme analizi arasındaki bağlantıyı ele almak ve bunların arasındaki ilişkiyi Ek I’deki ilgili gerekliliklere uygunluğun gösterilmesi için değerlendirmek, - Ek IX’un 5.2 ila 5.4 numaralı maddelerinde atıfta bulunulan spesifik prosedürleri yürütmek, - Sınıf IIa veya sınıf IIb cihazlar söz konusu olduğunda, temsili bir temelde cihazların teknik dokümantasyonunu değerlendirmek, - Uygun gözetim denetimlerini ve değerlendirmelerini planlamak ve periyodik olarak yürütmek, kalite yönetim sisteminin düzgün işleyişini doğrulamak için belirli testleri yürütmek veya talep etmek ve habersiz yerinde denetimler gerçekleştirmek, - Cihazların örneklenmesiyle ilgili olarak, imal edilen cihazın teknik dokümantasyona uygun olduğunu doğrulamak ve örnekleme öncesinde ilgili örnekleme kriterlerini ve test prosedürünü tanımlamak, - Bir imalatçının ilgili eklere uygunluğunu değerlendirmek ve doğrulamak. Onaylanmış kuruluş, imalatçı uygun olduğunu iddia etmese dahi, ilgili olduğu hallerde, mevcut ortak spesifikasyonları, kılavuz ve iyi uygulama dokümanlarını ve uyumlaştırılmış standartları dikkate alır. 4.5.2. Kalite yönetim sistemi denetimi a) Bir onaylanmış kuruluş; kalite yönetim sistemi değerlendirmesinin bir bölümü olarak denetimden önce ve dokümante edilmiş prosedürleri uyarınca; - İlgili uygunluk değerlendirme eki uyarınca sunulan dokümantasyonu değerlendirir ve imalatçının kalite yönetim sisteminin kapsamının tümünü göstermek ve bu Yönetmeliğin gerekliliklerini karşılayıp karşılamadığını belirlemek üzere gereken faaliyetlerin sayısını ve sırasını açıkça tanımlayan bir denetim programı hazırlar, - Farklı imalat tesisleri arasındaki bağlantıları ve bunlar arasında sorumlulukların dağılımını tespit eder ve imalatçının ilgili tedarikçilerini ve/veya yüklenicilerini tanımlar ve bu tedarikçilerin veya yüklenicilerin ya da her ikisinin özel olarak denetimine yönelik gereksinimi değerlendirir, - Denetim programında belirtilen her bir denetim için; denetimin hedeflerini, kriterlerini ve kapsamını açıkça tanımlar ve denetimin kapsadığı cihazlara, teknolojilere ve süreçlere yönelik spesifik gereklilikleri yeterli bir şekilde ele alan ve göz önünde tutan bir denetim planı hazırlar, - Sınıf IIa ve sınıf IIb cihazlar için; imalatçının başvurusu kapsamında yer alan bu tür cihazların çeşitliliğini kapsayan Ek II ve Ek III’te atıfta bulunulduğu şekilde teknik dokümantasyonun değerlendirilmesine yönelik bir örnekleme planı hazırlar ve bunu güncel tutar. Bu plan, sertifikanın kapsadığı tüm cihaz gamının sertifikanın geçerlilik süresi boyunca örneklenmesini sağlar, - Münferit denetimler yürütmek üzere nitelikli ve yetkili personeli gereğine uygun bir şekilde seçer ve atar. Ekip üyelerinin şahsi rolleri, sorumlulukları ve yetkileri açıkça tanımlanır ve dokümante edilir. b) Onaylanmış kuruluş; hazırladığı denetim programına dayanarak ve dokümante edilmiş prosedürleri uyarınca: - Kalite yönetim sisteminin, kapsadığı cihazların, tasarımdan nihai kalite kontrole ve sürekli gözetime kadar her aşamada bu Yönetmeliğin ilgili hükümlerine uygunluğunu sağladığını doğrulamak üzere; imalatçının kalite yönetim sistemini denetler ve bu Yönetmelik gerekliliklerinin karşılanıp karşılanmadığını belirler, - İlgili teknik dokümantasyona dayanarak imalatçının ilgili uygunluk değerlendirme ekinde atıfta bulunulan gereklilikleri karşılayıp karşılamadığını belirlemek üzere imalatçının süreçlerini ve alt sistemlerini özellikle: Tasarım ve geliştirme, Üretim ve süreç kontrolleri, Ürün dokümantasyonu, Satın alınan cihazların doğrulanması dâhil satın alma kontrolleri, Piyasaya arz sonrası gözetime yönelik olanlar dâhil düzeltici ve önleyici faaliyetler ve Piyasaya arz sonrası klinik takip yönünden inceler ve denetler, ayrıca Ek I’de belirtilen genel güvenlilik ve performans gerekliliklerini yerine getirmekle ilgili olanlar dâhil olmak üzere imalatçı tarafından benimsenen gereklilikleri ve hükümleri inceler ve denetler. Dokümantasyon; cihazın amaçlanan kullanımı, imalat teknolojilerinin karmaşıklığı, üretilen cihazların çeşitliliği ve sınıfları ve mevcut piyasaya arz sonrası gözetim bilgileri ile ilişkili riskleri yansıtacak şekilde örneklendirilir, - Hâlihazırda denetim programı kapsamında değilse, bitmiş cihazların uygunluğunun tedarikçilerin faaliyetlerinden önemli ölçüde etkilendiği durumda ve özellikle imalatçının tedarikçileri üzerinde yeterli kontrolü olduğunu gösteremediği durumda, imalatçının tedarikçilerinin tesislerinde süreçlerin kontrolünü denetler, - Klinik öncesi ve klinik değerlendirmeler için kendi örnekleme planına dayanarak ve bu Ekin 4.5.4 ile 4.5.5 numaralı maddeleri dikkate alarak teknik dokümantasyon değerlendirmelerini yürütür, - Onaylanmış kuruluş, denetim bulgularının, bu Yönetmelik gereklilikleri ve ilgili standartlar ya da MDCG tarafından geliştirilen veya kabul edilen iyi uygulama dokümanları uyarınca uygun ve tutarlı bir şekilde sınıflandırılmasını sağlar. 4.5.3. Ürün doğrulama Teknik dokümantasyonun değerlendirilmesi Onaylanmış kuruluşlar, Ek IX’un II. Bölümü uyarınca yapılan teknik dokümantasyon değerlendirmesi için aşağıdakiler hakkında yeterli uzmanlığa, tesislere ve dokümante edilmiş prosedürlere sahip olur: - Cihazın kullanımı, biyouyumluluk, klinik değerlendirme, risk yönetimi ve sterilizasyon gibi özel konuları incelemek üzere uygun nitelikli ve yetkili personelin tahsisi, - Tasarımın bu Yönetmeliğe uygunluğunun değerlendirilmesi ve bu Ekin 4.5.4 ila 4.5.6 numaralı maddelerinin dikkate alınması. Bu değerlendirme, imalatçılar tarafından giriş, süreç içi ve son kontrollerin uygulanmasının ve bu kontrollerin sonuçlarının incelenmesini içerir. Bu Yönetmeliğin gerekliliklerine uygunluğun değerlendirilmesi için ilave testlere veya başka kanıtlara gerek duyulursa, söz konusu onaylanmış kuruluş, cihazla ilgili yeterli fiziksel testleri veya laboratuvar testlerini yapar ya da imalatçıdan bu tür testleri yapmasını talep eder. Tip incelemeleri Onaylanmış kuruluş; aşağıdakileri yapma kapasitesi dâhil olmak üzere Ek X uyarınca cihazların tip incelemesi için dokümante edilmiş prosedürlere, yeterli uzmanlığa ve tesislere sahip olur: - Bu Ekin 4.5.4 ila 4.5.6 numaralı maddelerini dikkate alarak teknik dokümantasyonu inceleme ve değerlendirme ve tipin bu dokümantasyona uygun olarak imal edildiğini doğrulama, - Onaylanmış kuruluş tarafından veya onun sorumluluğu altında test edilmesi gereken bütün ilgili ve kritik parametreleri tanımlayan bir test planı oluşturma, - Bu parametrelerin seçimine yönelik gerekçesini dokümante etme, - İmalatçı tarafından benimsenen çözümlerin, Ek I’de belirtilen genel güvenlilik ve performans gerekliliklerini karşıladığını doğrulamak için uygun incelemeleri ve testleri yürütme. Bu tür incelemeler ve testler, imalatçının kullanmayı seçtiği ilgili standartları gerçekten uyguladığını doğrulamak için gerekli bütün testleri içerir, - Doğrudan onaylanmış kuruluş tarafından yürütülmeyecekse, gerekli testlerin nerede yapılacağı konusunda başvuru sahibi ile anlaşma, - Test sonuçlarına yönelik tüm sorumluluğu üstlenme. İmalatçı tarafından sunulan test raporları, sadece yetkin ve imalatçıdan bağımsız olan uygunluk değerlendirme kuruluşları tarafından düzenlenmişse dikkate alınır. Her ürünün inceleme ve test yoluyla doğrulanması Onaylanmış kuruluş: a) Ek XI’in B Kısmı uyarınca her ürünün inceleme ve test yoluyla doğrulanmasına yönelik, dokümante edilmiş prosedürlere, yeterli uzmanlığa ve tesislere sahip olur, b) Aşağıdakileri yapmak için, onaylanmış kuruluş tarafından veya onun sorumluluğu altında test edilmesi gereken bütün ilgili ve kritik parametreleri tanımlayan bir test planı oluşturur: - Sınıf IIb cihazlar için; cihazın AB tip inceleme sertifikasında belirtilen tipe ve bu Yönetmeliğin söz konusu cihazlara uygulanan gerekliliklerine uygunluğunu doğrulamak, - Sınıf IIa cihazlar için; Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona ve bu Yönetmeliğin söz konusu cihazlara uygulanan gerekliliklerine uygunluğunu teyit etmek, c) (b) bendinde atıfta bulunulan parametrelerin seçimine yönelik gerekçesini dokümante eder, ç) Ek XI’in 15 numaralı maddesinde belirtildiği şekilde her ürünü inceleyerek ve test ederek, cihazın bu Yönetmeliğin gerekliliklerine uygunluğunu doğrulamak için, uygun değerlendirmeleri ve testleri yürütmek üzere dokümante edilmiş prosedürlere sahip olur, d) Onaylanmış kuruluşun kendisi tarafından yürütülmeyecek gerekli testlerin ne zaman ve nerede gerçekleştirileceğine ilişkin başvuru sahibi ile bir anlaşmaya varmayı sağlayan dokümante edilmiş prosedürlere sahip olur, e) Dokümante edilmiş prosedürler uyarınca test sonuçlarına yönelik tüm sorumluluğu üstlenir. İmalatçı tarafından sunulan test raporları, sadece yetkin ve imalatçıdan bağımsız olan uygunluk değerlendirme kuruluşları tarafından düzenlenmişse dikkate alınır. 4.5.4. Klinik öncesi değerlendirmenin değerlendirilmesi Onaylanmış kuruluş; klinik öncesi hususların değerlendirilmesiyle ilgili, imalatçının prosedürlerinin ve dokümantasyonunun incelenmesine yönelik dokümante edilmiş işleyen prosedürlere sahip olur. Onaylanmış kuruluş, imalatçının prosedürlerinin ve dokümantasyonunun aşağıdakileri yeterli bir şekilde ele aldığını inceler, geçerli kılar ve doğrular: a) Klinik öncesi değerlendirmeyi ve özellikle: - Bilimsel klinik öncesi literatür taramasının ve - Laboratuvar testleri, simüle edilmiş kullanım testleri, bilgisayar modellemeleri, hayvan modellerinin kullanımı gibi klinik öncesi testlerin planlanmasını, yürütülmesini, değerlendirilmesini, raporlanmasını ve uygun olduğu hallerde güncellenmesini, b) Vücut temasının niteliği ve süresi ile birlikte ilişkili spesifik biyolojik riskleri, c) Risk yönetimi süreciyle bağlantıyı, ç) Ek I’in ilgili gerekliliklerine uygunluğu göstermek için mevcut klinik öncesi verilerin ve bunların uygunluğunun değerlendirilmesini ve analizini. Klinik öncesi değerlendirme prosedürleri ve dokümantasyonu ile ilgili onaylanmış kuruluşun değerlendirmesi; literatür taramalarının sonuçları ile birlikte gerçekleştirilen tüm validasyonları, doğrulamaları, testleri ve elde edilen sonuçları ele alır ve genellikle alternatif materyallerin ve maddelerin kullanımını göz önünde bulundurmayı içerir, ayrıca bitmiş cihazın ambalajını, raf ömrü dâhil stabilitesini dikkate alır. İmalatçı tarafından yeni bir test yapılmamış olması durumunda ya da prosedürlerden sapmalar olması durumunda, söz konusu onaylanmış kuruluş, imalatçı tarafından sunulan gerekçeyi kritik bir şekilde inceler. 4.5.5. Klinik değerlendirmenin değerlendirilmesi Onaylanmış kuruluş, hem ilk uygunluk değerlendirmesine yönelik hem de sürekli bir temelde, imalatçının klinik değerlendirmeye ilişkin prosedürlerinin ve dokümantasyonunun değerlendirilmesi ile ilgili olarak dokümante edilmiş işleyen prosedürlere sahip olur. Onaylanmış kuruluş, imalatçıların prosedürlerinin ve dokümantasyonlarının aşağıdakileri uygun bir şekilde ele aldığını inceler, geçerli kılar ve doğrular: - Ek XIV’te atıfta bulunulduğu şekilde, klinik değerlendirmenin planlanmasını, yürütülmesini, değerlendirilmesini, raporlanmasını ve güncellenmesini, - Piyasaya arz sonrası gözetimi ve piyasaya arz sonrası klinik takibi, - Risk yönetimi süreciyle bağlantıyı, - Ek I’in ilgili gerekliliklerine uygunluğunu göstermek için mevcut verinin ve bunun uygunluğunun değerlendirilmesini ve analizini, - Klinik kanıtlarla ilgili elde edilen sonuçlar ve klinik değerlendirme raporunun hazırlanmasını. Birinci paragrafta atıfta bulunulan bu prosedürler, mevcut ortak spesifikasyonları, kılavuz ve iyi uygulama dokümanlarını göz önünde bulundurur. Ek XIV’te atıfta bulunulduğu şekilde, onaylanmış kuruluşun klinik değerlendirme değerlendirmesi, aşağıdakileri kapsar: - İmalatçı tarafından belirtilen kullanım amacını ve cihaza yönelik açıklanan iddiaları, - Klinik değerlendirmenin planlanmasını, - Literatür taramasına yönelik metodolojiyi, - Literatür taramasından elde edilen ilgili dokümantasyonu, - Klinik araştırmayı, - Diğer cihazlara ilişkin iddia edilen eşdeğerliliğin geçerliliğini, eşdeğerliliğin gösterimini, eşdeğer ve benzer cihazlardan alınan verinin uygunluğunu ve sonuçlarını, - Piyasaya arz sonrası gözetim ve piyasaya arz sonrası klinik takibi, - Klinik değerlendirme raporunu, - Klinik araştırmaların veya piyasaya arz sonrası klinik takibin yapılmamasına ilişkin gerekçeleri. Klinik değerlendirmeye dâhil edilen klinik araştırmalardan elde edilen klinik verilerle ilgili olarak söz konusu onaylanmış kuruluş, imalatçı tarafından elde edilen sonuçların, onaylanan klinik araştırma planı ışığında geçerli olmasını sağlar. Onaylanmış kuruluş; klinik değerlendirmenin, Ek I’de belirtilen ilgili güvenlilik ve performans gerekliliklerini yeterince ele almasını, risk yönetimi gereklilikleriyle uygun bir şekilde aynı eksende olmasını, Ek XIV uyarınca yürütülmesini ve cihazla ilgili sunulan bilgilere uygun bir şekilde yansıtılmasını sağlar. 4.5.6. Spesifik prosedürler Onaylanmış kuruluş, Ek IX’un 5 ve 6 numaralı maddesinde, Ek X’un 6 numaralı maddesinde ve Ek XI’in 16 numaralı maddesinde atıfta bulunulan prosedürlerden atanmış olduklarına yönelik dokümante edilmiş prosedürlere, yeterli uzmanlığa ve tesislere sahip olur. Hayvan Kaynaklı Dokular Kullanılarak İmal Edilen Tıbbi Cihazlara Dair Yönetmelikte atıfta bulunulduğu şekilde Bulaşıcı Süngerimsi Ensefalopati (TSE) duyarlı türlerden elde edilenler gibi, hayvan kaynaklı dokular veya hücreler ya da bunların türevleri kullanılarak imal edilen cihazlar söz konusu olduğunda, onaylanmış kuruluş, ilgili yetkili otorite için özet bir değerlendirme raporu hazırlamak da dâhil olmak üzere, söz konusu Yönetmelikte belirtilen gereklilikleri karşılayan dokümante edilmiş işleyen prosedürlere sahip olur. 4.6. Raporlama Onaylanmış kuruluş: - Değerlendirme sonuçlarının açık olması, bu Yönetmeliğin gerekliliklerine uygunluğunun gösterilmesi ve atama otoritelerindeki personel gibi değerlendirmeye dâhil olmayan kişilere bu tür uygunluk ile ilgili objektif kanıtlar sunabilmek için uygunluk değerlendirmesinin tüm aşamalarının dokümante edilmesini sağlar, - Kalite yönetim sistemi denetimleri için açıkça görülebilir bir denetim izi sağlamak üzere yeterli kayıtların mevcut olmasını sağlar, - Klinik değerlendirmenin değerlendirilmesi raporunda, klinik değerlendirme ile ilgili değerlendirmesinin sonuçlarını açıkça dokümante eder, - Her bir spesifik proje için, MDCG tarafından belirlenen asgari bir dizi ögeyi içeren standart bir formata dayanan ayrıntılı bir rapor sağlar. Onaylanmış kuruluşun raporu: - Değerlendirmesinin çıktısını açıkça dokümante eder ve imalatçının bu Yönetmeliğin gerekliliklerine uygunluğunun doğrulanmasından elde edilen net bir sonuç içerir, - Onaylanmış kuruluş tarafından yapılacak nihai inceleme ve alınacak nihai karar için bir tavsiyede bulunur ve bu tavsiye onaylanmış kuruluşta sorumlu personelden biri tarafından imzalanır, - Söz konusu imalatçıya sunulur. 4.7. Nihai inceleme Onaylanmış kuruluş, nihai bir karar almadan önce: - Spesifik projeler hakkında nihai inceleme ve karar alma için atanan personelin, uygun bir şekilde yetkilendirilmesini ve değerlendirmeleri yürüten personelden farklı olmasını sağlar, - Değerlendirme süresince not edilen uygunsuzlukların çözümlenmesi ile ilgili olanlar da dâhil olmak üzere karar almak için gerekli rapor/raporların ve destekleyici dokümantasyonun, başvuru kapsamı açısından tam ve yeterli olduğunu doğrular, - Bir sertifikanın düzenlenmesini engelleyen çözümlenmemiş uygunsuzlukların olup olmadığını doğrular. 4.8. Kararlar ve belgelendirmeler Onaylanmış kuruluş; sertifikaların düzenlenmesi, askıya alınması, kısıtlanması ve geri çekilmesi için sorumlulukların tahsisi ile ilgili olanlar dâhil olmak üzere karar almaya yönelik dokümante edilmiş prosedürlere sahip olur. Bu prosedürler, bu Yönetmeliğin Beşinci Kısım’ında belirtilen bildirim gerekliliklerini içerir. Bu prosedürler, söz konusu onaylanmış kuruluşun: - Değerlendirme dokümantasyonuna ve mevcut ilave bilgilere dayanarak, bu Yönetmeliğin gerekliliklerinin yerine getirilip getirilmediğine karar vermesine, - Klinik değerlendirme ve risk yönetimi ile ilgili değerlendirmesinin sonuçlarına dayanarak, piyasaya arz sonrası klinik takip planı dâhil olmak üzere piyasaya arz sonrası gözetim planının yeterli olup olmadığına karar vermesine, - Güncel klinik değerlendirmeye ilişkin onaylanmış kuruluş tarafından daha ayrıntılı incelemeye yönelik spesifik kilit noktalara karar vermesine, - Belgelendirme için spesifik şartlar veya hükümler tanımlamanın gerekip gerekmediğine karar vermesine, - Cihazın; yeniliği, risk sınıflandırması, klinik değerlendirmesi ve risk analizinden elde edilen sonuçlar temelinde, 5 yılı geçmeyen bir belgelendirme süresine karar vermesine, - Sorumlu personelden birinin imzasıyla onay dâhil olmak üzere karar alma ve onay aşamalarını açıkça dokümante etmesine, - Özellikle, bir sertifikayı imzalayan son kişinin, karar alıcı veya alıcılardan farklı olması ya da bu Ekin 3.2.7 numaralı maddesinde belirtilen gereklilikleri yerine getirmemesi durumunda kararların bildirimine yönelik sorumlulukları ve mekanizmaları açıkça dokümante etmesine, - Ek XII’de belirtilen asgari gereklilikler uyarınca, 5 yılı geçmeyen bir geçerlilik süresi için ve belgelendirmeyle ilişkili kısıtlamaların veya spesifik şartların bulunup bulunmadığını gösteren sertifika veya sertifikalar düzenlemesine, - Birden çok kuruluşu kapsayan sertifikalar değil, sadece başvuru sahibi için sertifika veya sertifikalar düzenlemesine, - Değerlendirmenin çıktısı ve son karar konusunda imalatçının bilgilendirilmesini ve bunların 57 nci maddede atıfta bulunulan elektronik sisteme girilmesini sağlamasına imkân verir. 4.9. Değişiklikler ve modifikasyonlar Onaylanmış kuruluş; imalatçıların bilgi verme yükümlülükleri ile aşağıda belirtilen hususlardaki değişikliklerin değerlendirilmesi ile ilgili olarak işleyen ve dokümante edilmiş prosedürlere ve imalatçılarla yapılan sözleşmeye dayalı düzenlemelere sahip olur: - Onaylanmış kalite yönetim sistemi veya sistemleri ya da kapsanan ürün yelpazesi, - Onaylanmış cihaz tasarımı, - Cihazın kullanım amacı ya da cihaza ilişkin iddialar, - Onaylanmış cihaz tipi, - Bir cihazın ihtiva ettiği veya bir cihazın imalatı için kullanılan ve bu Ekin 4.5.6 numaralı maddesi uyarınca spesifik prosedürlere tabi olan maddeler. Birinci paragrafta atıfta bulunulan prosedürler ve sözleşmeye dayalı düzenlemeler, aynı paragrafta atıfta bulunulan değişiklerin önemini kontrol etmeye yönelik tedbirler içerir. Söz konusu onaylanmış kuruluş dokümante edilmiş prosedürleri uyarınca: - İmalatçıların, birinci paragrafta atıfta bulunulduğu şekilde değişikliklere yönelik planları ve bu tür değişikliklere ilişkin ilgili bilgileri ön onay için sunmasını sağlar, - Önerilen değişiklikleri değerlendirir ve bu değişikliklerden sonra kalite yönetim sisteminin ya da cihaz tasarımının veya cihaz tipinin hala bu Yönetmeliğin gerekliliklerini karşılayıp karşılamadığını doğrular, - İmalatçıya kararını bildirir ve değerlendirmesinin gerekçeli sonuçlarını içeren bir rapor veya uygulanabilir olduğunda bir ek rapor sunar. 4.10. Gözetim faaliyetleri ve belgelendirme sonrası izleme Onaylanmış kuruluş, aşağıdakilere yönelik dokümante edilmiş prosedürlere sahip olur: - İmalatçılarla ilgili gözetim faaliyetlerinin nasıl ve ne zaman yürütüleceğini tanımlamaya yönelik. Bu prosedürler, imalatçıların ve uygulanabildiği hallerde ürün testlerini yürüten yüklenicilerin ve tedarikçilerin habersiz yerinde denetimlerine yönelik düzenlemeleri ve belirli aralıklarla klinik veri güncellemeleri gibi imalatçıları bağlayan ve belgelendirme kararlarıyla ilişkili olan şartlara uygunluğun izlenmesini içerir, - Atanma kapsamlarıyla ilişkili olarak, ilgili bilimsel ve klinik veri kaynaklarını ve piyasaya arz sonrası bilgileri taramaya yönelik. Bu tür bilgiler, gözetim faaliyetlerinin planlanmasında ve yürütülmesinde dikkate alınır, - 89 uncu maddenin ikinci fıkrası kapsamında erişim sağladıkları vijilans verilerini, mevcut sertifikaların geçerliliği üzerinde varsa etkisini değerlendirmek için gözden geçirmeye yönelik. Değerlendirme sonuçları ve alınan kararlar tüm ayrıntılarıyla dokümante edilir. Söz konusu onaylanmış kuruluş, bir imalatçıdan veya yetkili otoriteden vijilans vakaları hakkında bilgi aldığında aşağıdaki seçeneklerden hangisini uygulayacağına karar verir: - Vijilans vakasının verilen sertifika ile açık bir şekilde ilgili olmadığı durumda herhangi bir faaliyette bulunmamak, - Verilen sertifikanın risk altında olup olmadığını ya da yeterli düzeltici faaliyetin yapılıp yapılmadığını belirlemek için imalatçının ve yetkili otoritenin faaliyetlerini ve imalatçının araştırma sonuçlarını gözlemlemek, - Verilen sertifikanın risk altında olması muhtemel olduğunda, doküman incelemeleri, kısa süreli veya habersiz denetimler ve ürün test etme gibi olağandışı gözetim tedbirlerini gerçekleştirmek, - Gözetim denetimlerinin sıklığını arttırmak, - İmalatçının bir sonraki denetimi vesilesiyle spesifik ürünleri ve süreçleri incelemek veya - İlgili diğer tedbirleri almak. İmalatçıların gözetim denetimleriyle ilgili olarak onaylanmış kuruluş, aşağıdakileri yapmak için dokümante edilmiş prosedürlere sahip olur: - Bu Ekin 4.5 numaralı maddesindeki ilgili gereklilikler doğrultusunda planlanan ve yürütülen imalatçının gözetim denetimlerini en azından yıllık olarak yürütmek, - Vijilans, piyasaya arz sonrası gözetim ve PMCF konusunda; imalatçının dokümantasyonunun yeterli şekilde değerlendirilmesini ve bunlarla ilgili hükümlerin uygulanmasını sağlamak, - İmalatçının, onaylanmış kalite yönetim sistemini sürekli olarak uygulamasını sağlamak için denetimler süresince, önceden tanımlanmış örnekleme kriterlerine ve test prosedürlerine göre cihazları ve teknik dokümantasyonu örneklemek ve test etmek, - İmalatçının ilgili eklerde belirtilen dokümantasyon ve bilgilendirme yükümlülüklerine uymasını ve prosedürlerinin kalite yönetim sistemlerinin uygulanması ile ilgili iyi uygulamaları dikkate almasını sağlamak, - İmalatçının, kalite yönetim sistemini veya cihaz onaylarını yanıltıcı bir şekilde kullanmamasını sağlamak, - Kalite yönetim sisteminin bu Yönetmeliğin gerekliliklerine uymaya devam edip etmediğini belirlemek için yeterli bilgi toplamak, - Uygunsuzluk tespit edilirse imalatçıdan düzeltmeler, düzeltici faaliyetler ve uygulanabildiği hallerde önleyici faaliyetler talep etmek, - Gerektiğinde ilgili sertifikaya spesifik kısıtlamalar getirmek ya da sertifikayı askıya almak veya geri çekmek. Onaylanmış kuruluş; belgelendirmeye yönelik şartların bir bölümü olması durumunda: - İmalatçının piyasaya arz sonrası gözetimine, piyasaya arz sonrası klinik takibine ve cihazla tedavi edilen tıbbi durumla ilgili klinik literatüre ya da benzer cihazlarla ilgili klinik literatüre dayanarak klinik değerlendirmenin imalatçı tarafından güncellenen en son versiyonunda derinlemesine bir inceleme yürütür, - Derinlemesine incelemenin sonuçlarını açık bir şekilde dokümante eder ve imalatçıya spesifik endişeleri iletir veya spesifik şartlar getirir, - Klinik değerlendirmenin güncellenen en son versiyonunun, kullanım talimatına ve uygulanabildiği hallerde güvenlilik ve performans özetine uygun bir şekilde yansıtılmasını sağlar. 4.11. Yeniden belgelendirme Onaylanmış kuruluş, yeniden belgelendirme incelemeleri ve sertifikaların yenilenmesi ile ilgili dokümante edilmiş işleyen prosedürlere sahip olur. Onaylanmış kalite yönetim sistemlerinin veya AB teknik dokümantasyon değerlendirme sertifikalarının ya da AB tip inceleme sertifikalarının yeniden belgelendirilmesi, asgari olarak her 5 yılda bir gerçekleştirilir. Onaylanmış kuruluş, AB teknik dokümantasyon değerlendirme sertifikalarının ve AB tip inceleme sertifikalarının yenilenmesi ile ilgili dokümante edilmiş prosedürlere sahip olur. Bu prosedürler, söz konusu imalatçının aşağıdakiler dâhil olmak üzere cihaza ilişkin değişikliklerin ve bilimsel bulguların bir özetini sunmasını gerektirir: a) Henüz bildirilmemiş olan değişiklikler dâhil olmak üzere başlangıçta onaylanan cihazdaki bütün değişiklikler, b) Piyasaya arz sonrası gözetimden kazanılan deneyim, c) Risk yönetiminden kazanılan deneyim, ç) Ek I’de belirtilen genel güvenlilik ve performans gerekliliklerine uygunluğun kanıtının güncellenmesinden kazanılan deneyim, d) Klinik araştırmaların ve PMCF’nin sonuçları dâhil olmak üzere klinik değerlendirme incelemelerinden kazanılan deneyim, e) Gerekliliklerdeki, cihaz bileşenlerindeki ya da bilimsel veya düzenleyici çevredeki değişiklikler, f) Uygulanan veya yeni uyumlaştırılmış standartlardaki, ortak spesifikasyonlardaki ya da eşdeğer dokümanlardaki değişiklikler, g) Tıbbi, bilimsel ve teknik bilgilerdeki değişiklikler, örneğin: - Yeni tedaviler, - Test yöntemlerindeki değişiklikler, - Biyouyumluluklarına dair bulgular dâhil olmak üzere materyaller ve bileşenlerle ilgili yeni bilimsel bulgular, - Karşılaştırılabilir cihazlara ilişkin çalışmalardan kazanılan deneyim, - Kayıtlardan elde edilen veriler ve kayıtlar, - Karşılaştırılabilir cihazlarla yapılan klinik araştırmalardan kazanılan deneyim. Onaylanmış kuruluş, ikinci paragrafta atıfta bulunulan bilgileri değerlendirmek için dokümante edilmiş prosedürlere sahip olur ve imalatçıların klinik değerlendirme raporlarındaki gerekli güncellemeler dâhil olmak üzere önceki belgelendirmeden veya yeniden belgelendirmeden itibaren yapılan piyasaya arz sonrası gözetim ve PMCF faaliyetlerinden elde edilen klinik verilere özellikle dikkat eder. Onaylanmış kuruluş, yeniden belgelendirme kararı için ilk belgelendirme kararında kullandığı yöntem ve ilkelerin aynısını kullanır. Gerektiğinde, başvuru ve başvurunun incelenmesi gibi belgelendirmeye yönelik gerçekleştirilen aşamaları dikkate almak suretiyle yeniden belgelendirme için ayrı formlar oluşturulur.
____________________________________________
Ek VIII SINIFLANDIRMA KURALLARI
I. BÖLÜM SINIFLANDIRMA KURALLARINA ÖZEL TANIMLAR 1. KULLANIM SÜRESİ 1.1. Geçici: Normal koşullar altında 60 dakikadan az bir süre için devamlı kullanımın amaçlandığını ifade eder. 1.2. Kısa süreli: Normal koşullar altında 60 dakika ile 30 gün arasındaki bir süre için devamlı kullanımın amaçlandığını ifade eder. 1.3. Uzun süreli: Normal koşullar altında 30 günden fazla bir süre için devamlı kullanımın amaçlandığını ifade eder. 2. İNVAZİV VE AKTİF CİHAZLAR 2.1. Vücut açıklığı: Göz küresinin dış yüzeyi de dâhil vücuttaki herhangi bir doğal açıklık ya da stoma gibi herhangi bir kalıcı yapay açıklığı ifade eder. 2.2. Cerrahi invaziv cihaz: a) Bir cerrahi operasyonla veya cerrahi operasyonun bir parçası olarak, vücut açıklıklarının mukoz membranlarından geçenler de dâhil olmak üzere, vücut yüzeyinden geçerek vücut içerisine penetre olan invaziv cihazı ve b) Vücut açıklığından geçmenin haricinde başka bir yolla penetrasyon oluşturan cihazı ifade eder. 2.3. Tekrar kullanılabilir cerrahi alet: Bir aktif cihaza bağlantısı olmaksızın, kesme, delme, testereleme, kazıma, sıyırma, klempleme, çekme, klipsleme veya benzer prosedürlerde cerrahi kullanımı amaçlanan ve imalatçısının öngördüğü şekilde temizlik, dezenfeksiyon ve sterilizasyon gibi uygun prosedürler yürütüldükten sonra yeniden kullanılması amaçlanan aleti ifade eder. 2.4. Aktif terapötik cihaz: Bir hastalığın, yaralanmanın veya sakatlığın tedavisi ya da hafifletilmesi amacıyla, biyolojik fonksiyonları veya yapıları desteklemek, değiştirmek, ikame etmek veya eski haline getirmek için tek başına ya da başka cihazlarla birlikte kullanılan herhangi bir aktif cihazı ifade eder. 2.5. Tanılama ve izleme amaçlı aktif cihaz: Fizyolojik durumları, sağlık durumlarını, hastalıkları veya konjenital bozuklukları tespit etmeye, tanılamaya, izlemeye ya da tedavi etmeye yönelik bilgileri temin etmek için tek başına ya da başka cihazlarla birlikte kullanılan herhangi bir aktif cihazı ifade eder. 2.6. Merkezi dolaşım sistemi: Arteriae pulmonales, aorta ascendens, arcus aortae, bifurcatio aortae’ya kadar aorta descendens, arteriae coronariae, arteria carotis communis, arteria carotis externa, arteria carotis interna, arteriae cerebrales, truncus brachiocephalicus, venae cordis, venae pulmonales, vena cava superior ve vena cava inferioru içeren kan damarlarını ifade eder. 2.7. Merkezi sinir sistemi: Beyin, meninks ve omuriliği ifade eder. 2.8. Hasarlı deri veya hasarlı mukoz membran: Yara ya da bir patolojik değişiklik veya hastalık sonrasında değişiklik gösteren deri bölgesi veya mukoz membranı ifade eder.
II. BÖLÜM UYGULAMA KURALLARI 3.1. Sınıflandırma kurallarının uygulanması, cihazların kullanım amacını esas alır. 3.2. Söz konusu cihazın başka bir cihazla birlikte kullanılması amaçlanıyorsa, sınıflandırma kuralları, cihazların her birine ayrı ayrı uygulanır. Bir tıbbi cihazın aksesuarları, birlikte kullanıldıkları cihazdan ayrı olarak kendi başlarına sınıflandırılır. 3.3. Bir cihazı çalıştıran veya cihazın kullanımını etkileyen yazılım, cihaz ile aynı sınıfta yer alır. Yazılım; herhangi bir cihazdan bağımsızsa, kendi başına sınıflandırılır. 3.4. Cihaz, sadece veya özellikle, vücudun belirli bir bölümünde kullanılmayacak ise belirtilen en kritik kullanıma dayanılarak değerlendirilir ve sınıflandırılır. 3.5. Cihazın kullanım amacına dayanarak aynı cihaza birçok kural ya da aynı kural dâhilinde birçok alt kural uygulanıyorsa, en yüksek sınıflandırmayı sağlayan en katı kural ve alt kural uygulanır. 3.6. Bu Ekin 1’inde atıfta bulunulan süre hesaplanırken devamlı kullanım: a) Bir işlem sırasında, kullanıma geçici olarak ara verilmesine ya da temizlik veya dezenfeksiyon gibi amaçlar için cihazın geçici olarak kullanım dışı bırakılmasına bakılmaksızın, aynı cihazın kullanım süresinin tamamını (Kullanıma ara vermenin veya kullanım dışı bırakmanın geçici olup olmadığı, kullanıma ara verildiği ya da cihazın kullanım dışı bırakıldığı dönemin öncesindeki ve sonrasındaki kullanım süresiyle ilişkili olarak belirlenir), b) İmalatçı tarafından aynı tipte başka bir cihazla ivedilikle değiştirilmesi amaçlanan bir cihazın, toplam kullanımını ifade eder. 3.7. Bir cihaz, söz konusu hastalık veya duruma ilişkin tanıyı kendisi sağladığında ya da tanılamaya yönelik belirleyici bilgiler sağladığında, bu cihazın doğrudan tanılamaya olanak verdiği kabul edilir.
III. BÖLÜM SINIFLANDIRMA KURALLARI 4. İNVAZİV OLMAYAN CİHAZLAR 4.1. Kural 1 İnvaziv olmayan bütün cihazlar; aşağıda belirtilen kurallardan biri kapsamında olmadıkça sınıf I olarak sınıflandırılır. 4.2. Kural 2 Kanı, vücut sıvılarını, hücreleri veya dokuları, sıvıları veya gazları vücuda vermek, tatbik etmek veya infüze etmek amacıyla aktarım (channelling) ya da muhafaza için tasarlanan bütün invaziv olmayan cihazlar: - Sınıf IIa, sınıf IIb veya sınıf III aktif cihaza bağlanabiliyorsa sınıf IIa olarak sınıflandırılır, - Kan torbaları hariç olmak üzere, kan veya diğer vücut sıvılarının aktarımı veya muhafazası için ya da organları, organ parçalarını veya vücut hücrelerini ve dokularını muhafaza etmek için kullanımı amaçlanıyorsa sınıf IIa olarak sınıflandırılır. Kan torbaları sınıf IIb olarak sınıflandırılır. Diğer bütün durumlarda, bu tür cihazlar sınıf I olarak sınıflandırılır. 4.3. Kural 3 Vücuda implante edilmesi veya tatbik edilmesi amaçlanan insan doku veya hücrelerinin, kanın, diğer vücut sıvılarının ya da başka sıvıların biyolojik veya kimyasal bileşimini değiştirmek üzere tasarlanan tüm invaziv olmayan cihazlar sınıf IIb olarak sınıflandırılır. Ancak söz konusu işlem; filtrasyon, santrifüj veya gaz ya da ısı alışverişinden ibaretse bu cihazlar sınıf IIa olarak sınıflandırılır. İnsan vücudundan alınmış olan insan hücreleri, dokuları veya organlarına doğrudan temas ederek in vitro kullanılması amaçlanan ya da vücuda implante edilmesi veya tatbik edilmesinden önce insan embriyolarıyla in vitro kullanılması amaçlanan bir maddeden veya maddelerin karışımından oluşan tüm invaziv olmayan cihazlar, sınıf III olarak sınıflandırılır. 4.4. Kural 4 Hasarlı deri veya hasarlı mukoz membranla temas eden tüm invaziv olmayan cihazlar: - Eksüdaların kompresyonu veya emilimi için mekanik bir bariyer olarak kullanılması amaçlanıyorsa sınıf I olarak sınıflandırılır, - Özellikle dermis tabakasının veya mukoz membranın tahrip olduğu ve sadece sekonder iyileşmeyle düzelebilen deri hasarlarında kullanılması amaçlanıyorsa sınıf IIb olarak sınıflandırılır, - Özellikle hasarlı derinin veya mukoz membranın mikro çevresinin yönetimi amaçlanıyorsa sınıf IIa olarak sınıflandırılır ve - Diğer bütün durumlarda sınıf IIa olarak sınıflandırılır. Bu kural, hasarlı mukoz membrana temas eden invaziv cihazlara da uygulanır. 5. İNVAZİV CİHAZLAR 5.1. Kural 5 Cerrahi invaziv cihazlar hariç olmak üzere, aktif bir cihaza bağlanması amaçlanmayan ya da sınıf I aktif cihaza bağlanması amaçlanan, vücut açıklıklarına ilişkin tüm invaziv cihazlar: - Geçici kullanımı amaçlanıyorsa sınıf I olarak sınıflandırılır, - Kısa süreli kullanımı amaçlanıyorsa sınıf IIa olarak sınıflandırılır; ancak farinkse kadar olan ağız boşluğunda, kulak zarına kadar olan kulak kanalında veya nazal boşlukta kullanılanlar sınıf I olarak sınıflandırılır ve - Uzun süreli kullanımı amaçlanıyorsa sınıf IIb olarak sınıflandırılır; ancak farinkse kadar olan ağız boşluğunda, kulak zarına kadar olan kulak kanalında veya nazal boşlukta kullanılıp mukoz membran tarafından emilme ihtimali olmayanlar sınıf IIa olarak sınıflandırılır. Cerrahi invaziv cihazlar hariç olmak üzere, sınıf IIa, sınıf IIb veya sınıf III aktif cihaza bağlanması amaçlanan, vücut açıklıklarına ilişkin tüm invaziv cihazlar sınıf IIa olarak sınıflandırılır. 5.2. Kural 6 Geçici kullanımı amaçlanan tüm cerrahi invaziv cihazlar sınıf IIa olarak sınıflandırılır. Ancak bu cihazlar: - Özellikle kalp veya merkezi dolaşım sistemi ile ilgili bir defekti, vücudun bu bölümlerine doğrudan temas yoluyla; kontrol etme, tanılama, izleme veya düzeltme amaçlı ise sınıf III olarak sınıflandırılır, - Tekrar kullanılabilir cerrahi aletler ise sınıf I olarak sınıflandırılır, - Özellikle kalp veya merkezi dolaşım sistemine ya da merkezi sinir sistemine doğrudan temas ederek kullanımı amaçlanıyor ise sınıf III olarak sınıflandırılır, - İyonlaştırıcı radyasyon formunda enerji sağlaması amaçlanıyor ise sınıf IIb olarak sınıflandırılır, - Biyolojik bir etkiye sahip ise ya da tamamı veya büyük çoğunluğu absorbe ediliyor ise sınıf IIb olarak sınıflandırılır veya - Bir dağıtım sistemi vasıtasıyla tıbbi ürünleri tatbik etme amaçlı ise ve uygulama şekli dikkate alındığında tıbbi ürünün tatbiki potansiyel olarak tehlike oluşturuyor ise sınıf IIb olarak sınıflandırılır. 5.3 Kural 7 Kısa süreli kullanımı amaçlanan tüm cerrahi invaziv cihazlar sınıf IIa olarak sınıflandırılır. Ancak bu cihazlar: - Özellikle kalp veya merkezi dolaşım sistemi ile ilgili bir defekti, vücudun bu bölümlerine doğrudan temas yoluyla; kontrol etme, tanılama, izleme veya düzeltme amaçlı ise sınıf III olarak sınıflandırılır, - Özellikle kalp veya merkezi dolaşım sistemine ya da merkezi sinir sistemine doğrudan temas ederek kullanımı amaçlanıyor ise sınıf III olarak sınıflandırılır, - İyonlaştırıcı radyasyon formunda enerji sağlaması amaçlanıyor ise sınıf IIb olarak sınıflandırılır, - Biyolojik bir etkiye sahip ise ya da tamamı veya büyük çoğunluğu absorbe ediliyor ise sınıf III olarak sınıflandırılır, - Dişlere yerleştirilen cihazlar hariç olmak üzere, vücutta kimyasal değişime uğruyor ise sınıf IIb olarak sınıflandırılır veya - İlaçları tatbik etme amaçlı ise sınıf IIb olarak sınıflandırılır. 5.4. Kural 8 Bütün implante edilebilir cihazlar ve uzun süreli cerrahi invaziv cihazlar sınıf IIb olarak sınıflandırılır. Ancak bu cihazlar: - Dişlere yerleştirilme amaçlı ise sınıf IIa olarak sınıflandırılır, - Kalbe, merkezi dolaşım sistemine ya da merkezi sinir sistemine doğrudan temas ederek kullanımı amaçlanıyor ise sınıf III olarak sınıflandırılır, - Biyolojik bir etkiye sahip ise ya da tamamı veya büyük çoğunluğu absorbe ediliyor ise sınıf III olarak sınıflandırılır, - Dişlere yerleştirilen cihazlar hariç olmak üzere, vücutta kimyasal değişime uğruyor ise sınıf III olarak sınıflandırılır, - Tıbbi ürünleri tatbik etme amaçlı ise sınıf III olarak sınıflandırılır, - Aktif implante edilebilir cihazlar veya aksesuarları ise sınıf III olarak sınıflandırılır, - Meme implantları veya cerrahi meşler ise sınıf III olarak sınıflandırılır, - Vidalar, kamalar, plaklar ve aletler gibi yardımcı bileşenler hariç olmak üzere, total veya kısmi eklem replasmanı amaçlı ise sınıf III olarak sınıflandırılır veya - Vidalar, kamalar, plaklar ve aletler gibi bileşenler hariç olmak üzere, omurga disk replasmanı amaçlı implantlar ya da omurgaya temas eden implante edilebilir cihazlar ise sınıf III olarak sınıflandırılır. 6. AKTİF CİHAZLAR 6.1. Kural 9 Enerji vermesi veya enerji değişimi yapması amaçlanan tüm aktif terapötik cihazlar sınıf IIa olarak sınıflandırılır. Ancak bu cihazların karakteristikleri; enerjinin niteliği, yoğunluğu ve uygulama yeri dikkate alındığında potansiyel olarak tehlike oluşturacak şekilde insan vücuduna enerji verebilen ya da vücut ile enerji değişimi yapabilen türde ise sınıf IIb olarak sınıflandırılır. Sınıf IIb aktif terapötik cihazların performansını kontrol etmesi veya izlemesi amaçlanan ya da bu tür cihazların performansını doğrudan etkilemesi amaçlanan tüm aktif cihazlar sınıf IIb olarak sınıflandırılır. Terapötik amaçlı olarak iyonlaştırıcı radyasyon yayan tüm aktif cihazlar; bu tür cihazları kontrol eden veya izleyen ya da onların performansını doğrudan etkileyen cihazlar da dâhil, sınıf IIb olarak sınıflandırılır. Aktif implante edilebilir cihazların performansını kontrol etmesi, izlemesi veya doğrudan etkilemesi amaçlanan tüm aktif cihazlar sınıf III olarak sınıflandırılır. 6.2. Kural 10 Tanılama ve izlemeye yönelik aktif cihazlar: - İnsan vücudu tarafından absorbe edilecek enerjiyi sağlayacak şekilde tasarlanmışsa sınıf IIa olarak sınıflandırılır; ancak gözle görülebilir bir spektrumda hastanın vücudunu aydınlatması amaçlanan cihazlar sınıf I olarak sınıflandırılır, - Radyofarmasötiklerin in vivo dağılımını görüntülemek amaçlıysa sınıf IIa olarak sınıflandırılır, - Hayati fizyolojik süreçleri doğrudan tanılamaya veya izlemeye imkân vermek üzere tasarlanmışsa sınıf IIa olarak sınıflandırılır. Ancak özellikle hayati fizyolojik parametreleri izleme amaçlı olmaları ve bu parametrelerdeki varyasyonların; kardiyak performanstaki, solunumdaki ve merkezi sinir sistemi aktivitesindeki varyasyonlar gibi hasta için ani tehlikeyle sonuçlanabilecek nitelikte olması halinde ya da hastanın ani tehlikede olduğu klinik durumlarda tanılama amaçlı olmaları halinde, sınıf IIb olarak sınıflandırılır. Müdahaleli radyoloji cihazları ve bu tür cihazları kontrol eden veya izleyen ya da bunların performansını doğrudan etkileyen cihazlar da dâhil, tanısal veya terapötik radyoloji amacıyla iyonlaştırıcı radyasyon yaymayı amaçlayan aktif cihazlar sınıf IIb olarak sınıflandırılır. 6.3. Kural 11 Tanılama veya terapötik amaçlı kararlar almak için kullanılan bilgileri sağlayan yazılımlar sınıf IIa olarak sınıflandırılır. Ancak, bu kararlar: - Ölüm veya kişinin sağlık durumunda geri dönüşü olmayan bir bozulmaya neden olabilecek bir etkiye sahip ise, bu yazılımlar sınıf III olarak sınıflandırılır, - Bir kişinin sağlık durumunda ciddi bozulma veya cerrahi bir müdahaleye neden olabilecek bir etkiye sahip ise, bu yazılımlar sınıf IIb olarak sınıflandırılır. Fizyolojik süreçleri izlemesi amaçlanan yazılımlar, sınıf IIa olarak sınıflandırılır. Ancak, hayati fizyolojik parametrelerin izlenmesi amaçlanıyorsa ve bu parametrelerdeki varyasyonlar hastada ani tehlike ile sonuçlanabilecek nitelikteyse, bu yazılımlar sınıf IIb olarak sınıflandırılır. Diğer tüm yazılımlar sınıf I olarak sınıflandırılır. 6.4. Kural 12 Tıbbi ürünleri, vücut sıvılarını veya diğer maddeleri vücuda tatbik etme ve/veya vücuttan uzaklaştırma amaçlı tüm aktif cihazlar sınıf IIa olarak sınıflandırılır. Ancak bu işlem, söz konusu maddelerin niteliği, ilgili vücut bölümü ve uygulama şekli dikkate alındığında potansiyel olarak tehlike oluşturacak şekildeyse sınıf IIb olarak sınıflandırılır. 6.5. Kural 13 Ek XVI’nın 4. numaralı maddesinde atıfta bulunulduğu şekilde yağ dokusunu azaltmak, uzaklaştırmak veya parçalamak için kullanılması amaçlanan ekipman, sınıf IIb olarak sınıflandırılır. Ek XVI’nın 5. numaralı maddesinde atıfta bulunulduğu şekilde cilt uygulamalarına yönelik insan vücudu üzerinde kullanılması amaçlanan yüksek yoğunluklu elektromanyetik radyasyon yayan ekipman sınıf IIb olarak sınıflandırılır. Ancak bu ekipman yalnızca tüy alma amaçlı ise sınıf IIa olarak sınıflandırılır. Ek XVI’nın 6. numaralı maddesinde atıfta bulunulduğu şekilde beyindeki sinirsel aktiviteyi değiştirmek için kafatasına penetre olan elektrik akımları ya da manyetik veya elektromanyetik alanlar uygulayan beyin stimülasyonu amaçlı ekipman, sınıf III olarak sınıflandırılır. Diğer aktif cihazların tümü sınıf I olarak sınıflandırılır. 7. ÖZEL KURALLAR 7.1. Kural 14 Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin 4 üncü maddesinin (l) bendinde tanımlandığı şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün dâhil olmak üzere ayrı olarak kullanıldığında aynı Yönetmeliğin 4 üncü maddesinin (b) bendinde belirtildiği şekilde tıbbi ürün olarak kabul edilebilen ve cihazların fonksiyonuna yardımcı olan bir maddeyi bütünleşik bir parça olarak ihtiva eden tüm cihazlar, sınıf III olarak sınıflandırılır. 7.2. Kural 15 Doğum kontrolü veya cinsel yolla bulaşan hastalıkların bulaşmasının önlenmesi için kullanılan tüm cihazlar sınıf IIb olarak sınıflandırılır. Ancak, bunlar implante edilebilir veya uzun süreli invaziv cihazlar ise sınıf III olarak sınıflandırılır. 7.3. Kural 16 Özellikle kontak lensleri dezenfekte etmek, temizlemek, durulamak veya uygun olduğu yerde nemlendirmek için kullanılması amaçlanan tüm cihazlar sınıf IIb olarak sınıflandırılır. Özellikle tıbbi cihazları dezenfekte etmek veya sterilize etmek için kullanılması amaçlanan tüm cihazlar, sınıf IIa olarak sınıflandırılır. Ancak, bu cihazlar, özellikle invaziv cihazları işlemin son noktası olarak dezenfekte etmeyi amaçlayan dezenfekte edici solüsyonlar veya yıkayıcı dezenfektanlar ise sınıf IIb olarak sınıflandırılır. Bu kural, kontak lensler hariç olmak üzere cihazları yalnızca fiziksel etki yoluyla temizlemesi amaçlanan cihazlara uygulanmaz. 7.4. Kural 17 Özellikle X-ışınıyla oluşturulan tanısal görüntüleri kaydetmesi amaçlanan cihazlar sınıf IIa olarak sınıflandırılır. 7.5. Kural 18 Cansız veya cansız hale getirilmiş hayvan kaynaklı dokular veya hücreler ya da bunların türevleri kullanılarak imal edilen ve yalnızca sağlam deriye temas etmesi amaçlanan cihazlar hariç olmak üzere; cansız veya cansız hale getirilmiş insan veya hayvan kaynaklı dokular veya hücreler ya da bunların türevleri kullanılarak imal edilen tüm cihazlar sınıf III olarak sınıflandırılır. 7.6. Kural 19 Nanomateryal ihtiva eden veya nanomateryalden oluşan tüm cihazlar: - Yüksek veya orta iç maruziyet potansiyeline sahip ise sınıf III olarak sınıflandırılır, - Düşük iç maruziyet potansiyeline sahip ise sınıf IIb olarak sınıflandırılır ve - Göz ardı edilebilir bir iç maruziyet potansiyeline sahip ise sınıf IIa olarak sınıflandırılır. 7.7. Kural 20 Cerrahi invaziv cihazlar hariç olmak üzere; tıbbi ürünleri inhalasyon yoluyla uygulama amaçlı, vücut açıklıklarına ilişkin tüm invaziv cihazlar sınıf IIa olarak sınıflandırılır. Ancak, bu cihazların etki mekanizması, uygulanan tıbbi ürünün etkililiği ve güvenliği üzerinde önemli bir etkiye sahipse ya da yaşamı tehdit eden durumları tedavi etmeleri amaçlanıyorsa, bu cihazlar sınıf IIb olarak sınıflandırılır. 7.8. Kural 21 Deriye uygulanması ya da vücut açıklığı yoluyla insan vücuduna girmesi amaçlanan ve insan vücudu tarafından absorbe edilen veya insan vücudunda lokal olarak dağılan maddelerden ya da maddelerin kombinasyonlarından oluşan cihazlar: - Kullanım amacını gerçekleştirmek üzere, bu cihazlar veya bu cihazların metabolizma ürünleri insan vücudu tarafından sistemik olarak absorbe ediliyorsa sınıf III olarak sınıflandırılır, - Kullanım amacını midede veya alt gastrointestinal kanalda gerçekleştiriyorsa ve bu cihazlar veya bu cihazların metabolizma ürünleri, insan vücudu tarafından sistemik olarak absorbe ediliyorsa sınıf III olarak sınıflandırılır, - Deriye uygulanıyorsa ya da nazal boşlukta veya farinkse kadar olan ağız boşluğunda uygulanıyor ve kullanım amaçlarını bu boşluklarda gerçekleştiriyorsa, sınıf IIa olarak sınıflandırılır ve - Diğer tüm durumlarda, sınıf IIb olarak sınıflandırılır. 7.9. Kural 22 Kapalı devre sistemler veya otomatik harici defibrilatörler gibi, cihaz tarafından gerçekleştirilen hasta yönetimini önemli ölçüde belirleyen bütünleşik veya birleşik diyagnostik fonksiyona sahip aktif terapötik cihazlar sınıf III olarak sınıflandırılır.
______________________________________________
Ek IX KALİTE YÖNETİM SİSTEMİNE VE TEKNİK DOKÜMANTASYONUN DEĞERLENDİRİLMESİNE DAYALI UYGUNLUK DEĞERLENDİRMESİ
I. BÖLÜM KALİTE YÖNETİM SİSTEMİ 1.İmalatçı, 10 uncu maddenin dokuzuncu fıkrasında tanımlandığı şekilde bir kalite yönetim sistemi oluşturur, dokümante eder ve uygular ve ilgili cihazların yaşam döngüsü boyunca bu sistemin etkinliğini sürdürür. İmalatçı; bu Ekin 2 numaralı maddesinde belirtildiği şekilde kalite yönetim sisteminin uygulanmasını sağlar ve 2.3 ile 2.4 numaralı maddelerinde belirlendiği şekilde denetime ve 3 numaralı maddesinde belirtildiği şekilde gözetime tabi olur. 2. Kalite yönetim sisteminin değerlendirilmesi 2.1. İmalatçı, kalite yönetim sisteminin değerlendirilmesi için bir onaylanmış kuruluşa aşağıdakileri içeren bir başvuru yapar: - İmalatçının adı ve kayıtlı işyerinin adresi ve kalite yönetim sistemi kapsamındaki ilave her türlü imalat yeri; imalatçının başvurusu, yetkili temsilcisi tarafından yapılıyorsa, yetkili temsilcinin adı ve kayıtlı işyerinin adresi, - Kalite yönetim sistemi kapsamındaki cihaz veya cihaz grubuna ilişkin ilgili tüm bilgiler, - Aynı cihazla ilgili kalite yönetim sistemi için başka bir onaylanmış kuruluşa başvuru yapılmadığına dair yazılı bir beyan ya da aynı cihazla ilgili kalite yönetim sistemine yönelik önceki başvurular hakkında bilgiler, - Uygunluk değerlendirme prosedürü kapsamındaki cihaz modeli için, 19 uncu madde ve Ek IV uyarınca bir AB uygunluk beyanı taslağı, - İmalatçının kalite yönetim sistemine ilişkin dokümantasyon, - Kalite yönetim sisteminden kaynaklanan ve bu Yönetmelik kapsamında gerekli olan yükümlülükleri yerine getirmek için işleyen prosedürlerin dokümante edilmiş açıklaması ve bu prosedürleri uygulamak için söz konusu imalatçının taahhüdü, - Kalite yönetim sisteminin yeterli ve etkili kalmasını sağlamak için işleyen prosedürlerin açıklaması ve bu prosedürleri uygulamak için imalatçının taahhüdü, - İmalatçının piyasaya arz sonrası gözetim sistemine ve uygulanabildiği hallerde PMCF planına ilişkin dokümantasyon ve 85 ila 89 uncu maddelerde belirtilen vijilansa ilişkin hükümlerden kaynaklanan yükümlülüklere ve Komisyonca kabul edilen ilgili tasarruflara uygunluğu sağlamak için oluşturulan prosedürler, - Piyasaya arz sonrası gözetim sistemini ve uygulanabildiği hallerde PMCF planını güncel tutmak için işleyen prosedürlerin tanımı; 85 ila 89 uncu maddelerde belirtilen vijilansa ilişkin hükümlerden kaynaklanan yükümlülüklere ve Komisyonca kabul edilen ilgili tasarruflara uygunluğu sağlayan prosedürler; bu prosedürleri uygulamak için imalatçının taahhüdü, - Klinik değerlendirme planına ilişkin dokümantasyon ve - En son teknolojik gelişmeleri dikkate alarak klinik değerlendirme planını güncel tutmak için mevcut prosedürlerin açıklaması. 2.2. Kalite yönetim sisteminin uygulanması, bu Yönetmeliğe uygunluğu sağlar. İmalatçı tarafından kalite yönetim sistemine yönelik benimsenen tüm unsurlar, gereklilikler ve hükümler; kalite el kitabı ile kalite programları, kalite planları ve kalite kayıtları gibi yazılı politikalar ve prosedürler biçiminde, sistematik ve düzenli bir şekilde dokümante edilir. Bununla birlikte, kalite yönetim sisteminin değerlendirilmesi için sunulacak dokümantasyon özellikle aşağıdakileri yeterli bir şekilde tanımlar: a) İmalatçının kalite hedeflerini, b) İşletmenin organizasyonunu ve özellikle: - Kritik prosedürlere ilişkin personel sorumluluklarının atanması ile ilgili organizasyonel yapıları, idari personelin sorumlulukları ile organizasyonel yetkilerini, - Kalite yönetim sisteminin işleyişinin etkili olup olmadığını ve özellikle uygun olmayan cihazların kontrolü dâhil bu sistemin istenilen tasarımı ve cihaz kalitesini gerçekleştirme kabiliyetini izleme yöntemlerini, - Cihazların tasarımının, imalatının ve/veya nihai doğrulama ve testinin ya da bu süreçlerin herhangi bir bölümünün başka bir tarafça yürütülmesi durumunda; özellikle bunlara uygulanan kontrol tipi ve kapsamını ve kalite yönetim sisteminin etkili işleyişini izleme yöntemlerini, - Gerektiğinde, bir yetkili temsilcinin atanmasına yönelik taslak yetki belgesi ve bu yetki belgesini kabul etmek için yetkili temsilciden alınan niyet mektubunu; c) Cihazların tasarımını izlemeye, doğrulamaya, valide etmeye ve kontrol etmeye yönelik prosedürleri ve teknikleri, bu prosedürlerden ve tekniklerden elde edilen veriler ve kayıtlarla birlikte ilgili dokümantasyonu. Bu prosedürler ve teknikler, özellikle aşağıdakileri kapsar: - İlgili yasal gerekliliklerin, yeterliliğin, sınıflandırmanın, eşdeğerliliğin ele alınmasının, uygunluk değerlendirme prosedürlerinin seçiminin ve bu prosedürlere uygunluğun tanımlanmasına yönelik süreçler dâhil, mevzuata uygunluğa yönelik stratejiyi, - Uygulanabilir ortak spesifikasyonları ve tercih edilmesi durumunda, uyumlaştırılmış standartları veya diğer yeterli çözümleri dikkate alarak, uygulanabilir genel güvenlilik ve performans gerekliliklerinin ve bu gereklilikleri yerine getirmeye yönelik çözümlerin tanımlanmasını, - Ek I’in 3 numaralı maddesinde atıfta bulunulduğu şekilde risk yönetimini, - PMCF dâhil, 61 inci madde ve Ek XIV uyarınca klinik değerlendirmeyi, - Uygun klinik öncesi değerlendirme dâhil, tasarım ve yapım ile ilgili uygulanabilir spesifik gereklilikleri, özellikle Ek I’in II. Bölümünün gerekliliklerini, yerine getirmeye yönelik çözümleri, - Cihazla birlikte temin edilecek bilgiler ile ilgili uygulanabilir spesifik gereklilikleri, özellikle Ek I’in III. Bölümünün gerekliliklerini yerine getirmeye yönelik çözümleri, - İmalatın her aşamasında çizimlerden, spesifikasyonlardan veya diğer ilgili dokümanlardan oluşturulan ve güncel tutulan cihaz tanımlama prosedürlerini ve - Tasarım veya kalite yönetim sistemi değişikliklerinin yönetimini. ç) İmalat aşamasındaki doğrulama ve kalite güvence tekniklerini, kullanılacak süreçleri ve prosedürleri (özellikle sterilizasyona ilişkin) ve ilgili dokümanları ve d) İmalat öncesinde, sırasında ve sonrasında yürütülecek uygun testleri ve denemeleri, bunların gerçekleştirilme sıklığını ve kullanılacak test ekipmanını. Bu test ekipmanının kalibrasyonunu yeterli şekilde geriye doğru izlemek mümkün olur. Ayrıca imalatçı; Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona onaylanmış kuruluşun erişimini sağlar. 2.3. Denetim Onaylanmış kuruluş; bu Ekin 2.2 numaralı maddesinde atıfta bulunulan gereklilikleri karşılayıp karşılamadığını belirlemek amacıyla kalite yönetim sistemini denetler. İmalatçının kalite yönetim sistemi ile ilgili olarak bir uyumlaştırılmış standart veya ortak spesifikasyon kullanması durumunda, onaylanmış kuruluş bu standartlara veya ortak spesifikasyonlara uygunluğu değerlendirir. Onaylanmış kuruluş; uyulmadığı usulünce gerekçelendirilmediği sürece, ilgili uyumlaştırılmış standartları veya ortak spesifikasyonları karşılayan bir kalite yönetim sisteminin bu standartların veya ortak spesifikasyonların kapsadığı gerekliliklere uyduğunu varsayar. Onaylanmış kuruluşun denetim ekibi; Ek VII’nin 4.3 ila 4.5 numaralı maddeleri uyarınca, ilgili teknolojinin değerlendirilmesinde geçmiş deneyimi olan en az bir üye içerir. Bu tür deneyimin kolayca anlaşılabilir veya uygulanabilir olmadığı durumlarda onaylanmış kuruluş, bu ekibin oluşumuna yönelik dokümante edilmiş bir gerekçe sunar. Değerlendirme prosedürü; imalat sürecini ve ilgili diğer süreçleri doğrulamak için imalatçının tesislerinde ve uygun görüldüğü takdirde, tedarikçilerinin ve/veya yüklenicilerinin tesislerinde denetimi içerir. Ayrıca, sınıf IIa ve sınıf IIb cihazlar söz konusu olduğunda, kalite yönetim sistemi değerlendirmesine, bu Ekin 4 numaralı maddesinde belirtildiği gibi temsili bir temelde seçilen cihazlara ait teknik dokümantasyonun değerlendirilmesi eşlik eder. Temsili örneklerin seçiminde, onaylanmış kuruluş; MDCG tarafından geliştirilen yayımlanmış kılavuzları ve özellikle teknolojinin yeniliğini, tasarımdaki benzerlikleri, teknolojiyi, imalat ve sterilizasyon yöntemlerini, kullanım amacını ve bu Yönetmelik uyarınca yürütülmüş olan örneğin, fiziksel, kimyasal, biyolojik veya klinik özelliklere ilişkin, önceki ilgili değerlendirmelerin sonuçlarını dikkate alır. Söz konusu onaylanmış kuruluş, alınan örneklere yönelik gerekçesini dokümante eder. Kalite yönetim sistemi bu Yönetmeliğin ilgili hükümlerini karşılıyorsa, onaylanmış kuruluş bir AB kalite yönetim sistemi sertifikası düzenler. Onaylanmış kuruluş, sertifika düzenleme kararını imalatçıya bildirir. Bu karar, denetim sonuçlarını ve gerekçeli bir raporu içerir. 2.4. Söz konusu imalatçı; kalite yönetim sisteminde ya da kapsadığı cihaz çeşitliliğinde önemli değişikliklere yönelik her planını, kalite yönetim sistemini onaylayan onaylanmış kuruluşa bildirir. Onaylanmış kuruluş; teklif edilen değişiklikleri değerlendirir, ilave denetimlere yönelik ihtiyacı belirler ve bu değişikliklerden sonra kalite yönetim sisteminin bu Ekin 2.2 numaralı maddesinde atıfta bulunulan gereklilikleri karşılamaya devam edip etmediğini doğrular. Onaylanmış kuruluş, değerlendirmenin sonuçlarını ve uygulanabildiği hallerde ilave denetimlerin sonuçlarını içeren kararını imalatçıya bildirir. Kalite yönetim sistemindeki veya kapsadığı cihaz çeşitliliğindeki önemli her değişikliğe ilişkin onay, AB kalite yönetim sistemi sertifikasına ek bir belge şeklinde olur. 3. 3. Gözetim değerlendirmesi 3.1. Gözetimin amacı, imalatçının, onaylanan kalite yönetim sisteminden doğan yükümlülüklerini usulünce yerine getirmesini sağlamaktır. 3.2. İmalatçı, yerinde denetimler dâhil gerekli tüm denetimleri yürütmesi için onaylanmış kuruluşa yetki verir ve özellikle aşağıdakiler olmak üzere ilgili tüm bilgileri bu kuruluşa temin eder: - Kalite yönetim sistemine ilişkin dokümantasyonu, - Cihazların temsili bir örneği için, PMCF planı dâhil piyasaya arz sonrası gözetim planının ve 85 ila 89 uncu maddelerde belirtilen vijilansa ilişkin hükümlerin ve Komisyonca kabul edilen ilgili tasarrufların uygulanmasından elde edilen bulgulara ve sonuçlara ilişkin dokümantasyonu, - Kalite yönetim sisteminin tasarımla ilgili bölümünde şart koşulan verileri, örneğin; analizlerin, hesaplamaların, testlerin sonuçları ve Ek I’in 4 numaralı maddesinde atıfta bulunulduğu şekilde risk yönetimiyle ilgili benimsenen çözümleri ve - Kalite yönetim sisteminin imalat ile ilgili bölümünde şart koşulan verileri, örneğin; kalite kontrol raporları ve test verileri, kalibrasyon verileri ve ilgili personelin yetkinliklerine ilişkin kayıtları. 3.3. Onaylanmış kuruluşlar; periyodik olarak her 12 ayda asgari bir kez, söz konusu imalatçının onaylanan kalite yönetim sistemini ve piyasaya arz sonrası gözetim planını uyguladığından emin olmak için uygun denetimleri ve değerlendirmeleri yürütür. Bu denetimler ve değerlendirmeler, imalatçının ve uygun görüldüğü takdirde, tedarikçilerinin ve/veya yüklenicilerinin tesislerindeki denetimleri içerir. Bu tür yerinde denetimler sırasında onaylanmış kuruluş, gerekli olduğu durumlarda, kalite yönetim sisteminin düzgün bir şekilde işlediğini kontrol etmek amacıyla testler yapar veya talep eder. Onaylanmış kuruluş, imalatçıya gözetim denetimi raporunu ve eğer test yapılmış ise test raporunu sunar. 3.4. Onaylanmış kuruluş; her 5 yılda asgari bir kez, bu Ekin 3.3 numaralı maddesinde atıfta bulunulan periyodik gözetim değerlendirmeleriyle birleştirilebilen ya da bu gözetim değerlendirmesine ilave olarak gerçekleştirilebilen, imalatçının ve uygun olduğu hallerde tedarikçilerinin ve/veya yüklenicilerinin tesislerinde rastgele habersiz denetimler gerçekleştirir. Onaylanmış kuruluş, bu tür habersiz yerinde denetimler için bir plan oluşturur; fakat bunu imalatçıya bildirmez. 52 nci maddenin yedinci fıkrasının ikinci cümlesinde atıfta bulunulan cihazlar hariç olmak üzere bu tür habersiz yerinde denetimler kapsamında, onaylanmış kuruluş, imal edilen cihazın teknik dokümantasyona uygun olduğunu doğrulamak için üretilen cihazların yeterli miktarda numunesini ya da imalat süreçlerinden alınan yeterli miktarda numuneyi test eder. Habersiz yerinde denetimlerden önce onaylanmış kuruluş, ilgili numune alma kriterlerini ve test prosedürünü belirler. 52 nci maddenin yedinci fıkrasının ikinci cümlesinde atıfta bulunulan cihazlar hariç olmak üzere, imal edilen cihazın teknik dokümantasyona uygun olduğunu doğrulamak için onaylanmış kuruluş; ikinci paragrafta atıfta bulunulduğu şekilde numune almanın yanı sıra piyasadaki cihazlardan numune alır ya da sadece piyasadaki cihazlardan numune alır. Söz konusu onaylanmış kuruluş, numune almadan önce, ilgili numune alma kriterlerini ve test prosedürünü belirler. Onaylanmış kuruluş, söz konusu imalatçıya, mevcutsa numune testinin sonucunu içeren, yerinde denetim raporunu sunar 3.5. Sınıf IIa ve sınıf IIb cihazlar söz konusu olduğunda gözetim değerlendirmesi; bu Ekin 2.3 numaralı maddesinin üçüncü paragrafı uyarınca onaylanmış kuruluş tarafından dokümante edilen gerekçeye göre seçilen ilave temsili örneklere dayanarak ilgili cihaz veya cihazlar için bu Ekin 4 numaralı maddesinde belirtildiği şekilde teknik dokümantasyonun bir değerlendirmesini de içerir. Sınıf III cihazlar söz konusu olduğunda gözetim değerlendirmesi; uygun olduğu hallerde, üretilen veya satın alınan parçaların ve/veya malzemelerin miktarlarının bitmiş cihazların miktarlarına karşılık geldiğinin kontrolü dâhil, cihazın bütünlüğü için esas olan onay verilmiş parçaların ve/veya malzemelerin testini de içerir. 3.6. Onaylanmış kuruluş, değerlendirme ekibinin; ilgili cihazların, sistemlerin ve süreçlerin değerlendirilmesine yönelik yeterli deneyime, sürekli objektifliğe ve tarafsızlığa sahip olacak şekilde oluşturulmasını ve değerlendirme ekibi üyelerinin uygun aralıklarla rotasyonunu sağlar. Genel bir kural olarak bir baş denetçi art arda 3 yıldan daha fazla aynı imalatçıyla ilgili denetimleri yönetemez veya bu denetimlere katılamaz. 3.7. Onaylanmış kuruluş, üretilen cihazlardan veya piyasadan alınan numune ile teknik dokümantasyonda belirtilen spesifikasyonlar veya onaylanan tasarım arasında bir farklılık bulursa ilgili sertifikayı askıya alır veya geri çeker ya da sertifikaya kısıtlamalar getirir.
II. BÖLÜM TEKNİK DOKÜMANTASYONUN DEĞERLENDİRİLMESİ 4. Sınıf III cihazlara ve 52 nci maddenin dördüncü fıkrasının (b) bendinde atıfta bulunulan sınıf IIb cihazlara uygulanabilir teknik dokümantasyon değerlendirmesi 4.1. Bu Ekin 2 numaralı maddesinde belirtilen yükümlülüklere ek olarak imalatçı, piyasaya arz etmeyi ya da hizmete sunmayı planladığı ve kalite yönetim sisteminin kapsadığı cihazla ilgili teknik dokümantasyonun değerlendirilmesi için onaylanmış kuruluşa bir başvuruda bulunur. 4.2. Başvuru; söz konusu cihazın tasarımını, imalatını ve performansını tanımlar. Bu başvuru, Ek II ve Ek III’te atıfta bulunulduğu şekilde teknik dokümantasyonu içerir. 4.3. Onaylanmış kuruluş, ilgili teknolojinin, cihazların ve klinik kanıtların değerlendirilmesinde kanıtlanmış bilgi ve deneyime sahip personel aracılığıyla teknik dokümantasyonu değerlendirir. Onaylanmış kuruluş, bu Yönetmeliğin ilgili gerekliliklerine uygunluğun değerlendirilmesine imkân sağlamak üzere, ilave testler yaptırarak ya da daha fazla kanıt isteyerek başvurunun tamamlanmasını talep edebilir. Onaylanmış kuruluş, cihazla ilgili yeterli fiziksel testleri veya laboratuvar testlerini yapar ya da imalatçıdan bu tür testleri yapmasını talep eder. 4.4. Onaylanmış kuruluş, imalatçı tarafından klinik değerlendirme raporunda ve yapılan ilgili klinik değerlendirmede sunulan klinik kanıtı inceler. Onaylanmış kuruluş; yeterli klinik uzmanlığa sahip cihaz inceleyicileri istihdam eder ve gerektiği takdirde bu inceleme amaçları doğrultusunda, söz konusu cihazla ya da cihazın kullanıldığı klinik durumla ilgili doğrudan ve güncel deneyime sahip dış klinik uzmanlara başvurur. 4.5. Onaylanmış kuruluş; klinik kanıtların, değerlendirme kapsamındaki cihaza eşdeğer olduğu iddia edilen cihazlardan elde edilen verilere kısmen veya tamamen dayandırıldığı durumlarda, yeni endikasyonlar ve inovasyon gibi faktörleri dikkate alarak bu tür verileri kullanmanın uygunluğunu değerlendirir. Onaylanmış kuruluş, iddia edilen eşdeğerlilik hakkındaki ve uygunluğu göstermeye yönelik verilerin ilgisi ve yeterliliği hakkındaki değerlendirmelerini açıkça dokümante eder. İmalatçı tarafından inovatif olduğu iddia edilen her cihaz karakteristiği için ya da yeni endikasyonlar için onaylanmış kuruluş, spesifik iddiaların belirli klinik öncesi ve klinik verilerle ve risk analiziyle hangi ölçüde desteklendiğini değerlendirir. 4.6. Onaylanmış kuruluş, klinik kanıtların ve klinik değerlendirmenin yeterli olduğunu ve ilgili genel güvenlilik ve performans gerekliliklerine uygunluk hakkında imalatçı tarafından varılan sonuçları doğrular. Bu doğrulama; fayda-risk belirlemesinin, risk yönetiminin, kullanım talimatlarının, kullanıcı eğitiminin ve imalatçının piyasaya arz sonrası gözetim planının yeterliliğinin değerlendirilmesini içerir ve uygulanabildiği hallerde, önerilen PMCF planına yönelik ihtiyaca ve bunun yeterliliğine ilişkin bir incelemeyi kapsar. 4.7. Onaylanmış kuruluş yaptığı klinik kanıt değerlendirmesine dayanarak; klinik değerlendirmeyi ve fayda-risk belirlemesini inceler ve piyasaya arz sonrası gözetim ve PMCF verilerinden elde edilen klinik kanıta yönelik güncellemeleri gözden geçirebilmesine imkân tanıyan spesifik kilit noktaların tanımlanmasının gerekip gerekmediğini değerlendirir. 4.8. Onaylanmış kuruluş; klinik değerlendirmenin değerlendirilmesi raporunda, değerlendirmesinin sonucunu açıkça dokümante eder. 4.9. Onaylanmış kuruluş; klinik değerlendirmenin değerlendirilmesi raporu da dâhil teknik dokümantasyon değerlendirmesine ilişkin bir raporu imalatçıya sunar. Cihaz bu Yönetmeliğin ilgili hükümlerine uyuyorsa, onaylanmış kuruluş bir AB teknik dokümantasyon değerlendirme sertifikası düzenler. Bu sertifika, teknik dokümantasyon değerlendirmesinin sonuçlarını, sertifikanın geçerlilik şartlarını, onaylanan tasarımının tanımlanması için gereken verileri ve uygun olduğu hallerde, cihazın kullanım amacının açıklamasını içerir. 4.10. Onaylanan cihazdaki değişiklikler; cihazın güvenlilik ve performansını ya da cihazın kullanımı için öngörülen şartları etkileyebilecekse, AB teknik dokümantasyon değerlendirme sertifikasını düzenlemiş olan onaylanmış kuruluştan onay gerektirir. İmalatçı; yukarıda bahsedilen değişikliklerden herhangi birini uygulamayı planlaması durumunda, AB teknik dokümantasyon değerlendirme sertifikasını düzenlemiş olan onaylanmış kuruluşu bunlarla ilgili bilgilendirir. Onaylanmış kuruluş, planlanan değişiklikleri değerlendirir ve bunların 52 nci madde uyarınca yeni bir uygunluk değerlendirmesi gerektirip gerektirmediğine ya da AB teknik dokümantasyon değerlendirme sertifikasına ek bir belge vasıtasıyla belirtilip belirtilemeyeceğine karar verir. Ek bir belge vasıtasıyla belirtilebileceği durumda, onaylanmış kuruluş değişiklikleri değerlendirir, kararını imalatçıya bildirir ve değişikliklerin onaylanması durumunda, AB teknik dokümantasyon değerlendirme sertifikasına ek bir belge düzenler. 5. Spesifik ilave prosedürler 5.1. Belirli sınıf III ve sınıf IIb cihazlar için değerlendirme prosedürü a) Sınıf III implante edilebilir cihazlar için ve Ek VIII’in 6.4 numaralı maddesinde (Kural 12) atıfta bulunulduğu şekilde bir tıbbi ürünü vücuda tatbik etmesi ve/veya vücuttan uzaklaştırması amaçlanan sınıf IIb aktif cihazlar için onaylanmış kuruluş; 61 inci maddenin on birinci fıkrasında atıfta bulunulan imalatçının klinik değerlendirme raporunu destekleyen klinik verilerin kalitesini doğruladıktan sonra özellikle fayda-risk belirlemesi ile ilgili olmak üzere imalatçı tarafından sağlanan klinik kanıtlara, bu kanıtların tıbbi endikasyon/endikasyonlar da dâhil kullanım amacıyla tutarlılığına ve 10 uncu maddenin üçüncü fıkrasında ve Ek XIV’ün B Kısmında atıfta bulunulan piyasaya arz sonrası klinik takip planına ilişkin değerlendirmelerini belirten bir klinik değerlendirmenin değerlendirilmesi raporu hazırlar. Onaylanmış kuruluş; klinik değerlendirmenin değerlendirilmesi raporunu, Ek II’nin 6.1 numaralı maddesinin (c) ve (ç) bentlerinde atıfta bulunulan imalatçının klinik değerlendirme dokümantasyonu ile birlikte, bilimsel, teknik ve klinik görüş ve tavsiyelerde bulunmak üzere Komisyon tarafından teşkil edilen uzman heyetine ivedilikle iletmek üzere Komisyon’a sunar. b) Onaylanmış kuruluştan, (a) bendinde belirtilen değerlendirmelerini ilgili uzman heyetine sunması talep edilebilir. c) Uzman heyeti; Komisyonun gözetimi altında, imalatçı tarafından sağlanan klinik kanıtlar temelinde, özellikle fayda-risk belirlemesine, bu kanıtların tıbbi endikasyon/endikasyonlarla tutarlılığına ve PMCF planına ilişkin onaylanmış kuruluşun klinik değerlendirmenin değerlendirilmesi raporu hakkında bilimsel bir görüş sunup sunmamaya aşağıdaki kriterlerin tümüne dayanarak karar verir: i) Cihazın veya dâhil olduğu ilgili klinik prosedürün yeniliği ve bunların olası önemli klinik veya sağlık etkisi, ii) Bileşenler veya kaynak materyal bakımından ya da cihazın bozulması durumunda sağlık üzerindeki etkisi bakımından bilimsel olarak geçerli sağlık endişeleri nedeniyle spesifik cihaz kategorisi veya grubunun fayda-risk profilinde önemli ölçüde advers değişiklik, iii) Spesifik bir cihaz kategorisi veya grubu bakımından 85 inci madde uyarınca raporlanan ciddi olumsuz olayların oranında önemli ölçüde artış. Bu bilimsel görüş, (a) bendinde atıfta bulunulduğu şekilde Komisyondan dokümanların alındığı günden başlayan 60 günlük süre içerisinde verilir. (i), (ii) ve (iii) alt bentlerindeki kriterlere dayanarak bilimsel bir görüş verme kararına yönelik gerekçeler, bilimsel görüşe dâhil edilir. Sunulan bilgiler, uzman heyetinin bir sonuca varması için yeterli değilse bu durum bilimsel görüşte belirtilir. ç) Uzman heyeti; Komisyonun gözetimi altında, (c) bendinde belirtilen kriterlere dayanarak bilimsel bir görüş vermemeye karar verebilir; bu durumda, en kısa süre içerisinde ve her koşulda Komisyondan (a) bendinde atıfta bulunulduğu şekilde belgelerin alınmasından itibaren 21 gün içerisinde onaylanmış kuruluşu bilgilendirir. Uzman heyeti, bu süre içerisinde, kararına yönelik gerekçelerini onaylanmış kuruluşa ve Komisyon’a sunar; bunu takiben onaylanmış kuruluş bu cihazın belgelendirme prosedürüne devam edebilir. d) Uzman heyeti, Komisyondan belgelerin alınmasından itibaren 21 gün içerisinde, (c) bendi uyarınca bilimsel bir görüş verme ya da (ç) bendi uyarınca bilimsel bir görüş vermeme düşüncesini EUDAMED aracılığıyla Komisyon’a bildirir. e) 60 günlük süre içerisinde hiçbir görüş verilmemesi durumunda, onaylanmış kuruluş söz konusu cihazın belgelendirme prosedürüne devam edebilir. f) Onaylanmış kuruluş, uzman heyetinin bilimsel görüşünde ifade edilen görüşlere gereken önemi gösterir. Uzman heyetinin klinik kanıt seviyesinin yetersiz olduğunu veya bunun dışında fayda-risk belirlemesine, bu kanıtın tıbbi endikasyon/endikasyonlar dâhil olmak üzere kullanım amacıyla tutarlılığına ve piyasaya arz sonrası klinik takip planına ilişkin ciddi endişelerin bulunduğunu tespit etmesi durumunda, onaylanmış kuruluş, gerektiği takdirde, cihazın kullanım amacını belirli hasta grupları veya belirli tıbbi endikasyonlar için kısıtlamayı ve/veya sertifikanın geçerlilik süresine sınır koymayı, spesifik piyasaya arz sonrası klinik takip çalışmaları yürütmeyi, güvenlilik ve performans özetini veya kullanım talimatını uyarlamayı ya da uygun görüldüğü takdirde, uygunluk değerlendirme raporuna başka kısıtlamalar koymayı imalatçıya tavsiye eder. Onaylanmış kuruluş, uzman heyetinin tavsiyesine uymaması durumunda, uygunluk değerlendirme raporunda tam bir gerekçe sunar. 103 üncü maddeye halel gelmeksizin, hem uzman heyetinin bilimsel görüşü hem de onaylanmış kuruluş tarafından sunulan yazılı gerekçe Komisyon tarafından EUDAMED vasıtasıyla kamuya açık hale getirilir 5.2. Tıbbi madde ihtiva eden cihazlar söz konusu olduğundaki prosedür a) İnsan kanından veya insan plazmasından elde edilen ve cihazın fonksiyonuna yardımcı olan bir tıbbi ürün dâhil ayrı olarak kullanıldığında Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin 4 üncü maddesinin (b) bendi kapsamında bir tıbbi ürün olarak kabul edilebilen bir maddeyi, bir cihazın bütünleşik bir parça olarak ihtiva etmesi durumunda, bu maddenin kalitesi, güvenliliği ve yararlılığı , aynı Yöntemelik’in Ek 1’inde belirtilen metotlarla analoji yapılarak doğrulanır. b) Onaylanmış kuruluş; bir AB teknik dokümantasyon değerlendirme sertifikası düzenlemeden önce cihazın bir parçası olarak maddenin yararlılığını doğrulayarak ve cihazın kullanım amacını dikkate alarak bundan sonra bu bent kapsamında hangisine danışıldığına bağlı olarak burada “tıbbi ürünler danışma otoritesi” adıyla atıfta bulunulacak olan Beşeri Tıbbi Ürünler alanında Kurum ve/veya AB üyesi ülkeler tarafından atanan yetkili otoritelerin birinden ya da EMA’dan, cihazın bu maddeyi ihtiva etmesinin fayda veya riskleri dâhil maddenin kalitesi ve güvenliliği hakkında bilimsel görüş ister. Cihazın, bir insan kanı veya plazma türevi ya da ayrı olarak kullanıldığında AB’nin ilgili mevzuatı temel alınarak Kurumca yayımlanan kılavuzlar kapsamında yer alan bir tıbbi ürün olduğu kabul edilebilen bir madde ihtiva etmesi durumunda, onaylanmış kuruluş EMA’nın görüşünü ister. c) Tıbbi ürünler danışma otoritesi; görüşünü verirken imalat sürecini ve cihazın maddeyi ihtiva etmesinin yararlılığına ilişkin onaylanmış kuruluş tarafından belirlenen verileri dikkate alır. ç) Tıbbi ürünler danışma otoritesi, gerekli bütün dokümantasyonu almasından itibaren 210 gün içerisinde, onaylanmış kuruluşa görüşünü sunar. d) Tıbbi ürünler danışma otoritesinin bilimsel görüşü ve bu görüşün olası güncellemeleri, onaylanmış kuruluşun cihaza ilişkin dokümantasyonuna dâhil edilir. Onaylanmış kuruluş, kararını verirken bilimsel görüşte ifade edilen görüşlere gereken önemi verir. Onaylanmış kuruluş, bilimsel görüş olumsuz ise sertifikayı vermez ve nihai kararını tıbbi ürünler danışma otoritesine iletir. e) Cihazın ihtiva ettiği yardımcı maddeyle, özellikle de imalat süreciyle ilgili olarak, herhangi bir değişiklik yapılmadan önce imalatçı, bu değişiklikleri onaylanmış kuruluşa bildirir. Bu onaylanmış kuruluş, yardımcı maddenin kalitesinin ve güvenliliğinin değişmediğini teyit etmek için tıbbi ürünler danışma otoritesinin görüşünü ister. Tıbbi ürünler danışma otoritesi; bu değişikliklerin, cihazın maddeyi ihtiva etmesiyle ilgili olarak önceden belirlenen risk veya fayda üzerinde olumsuz etkisinin olmadığından emin olmak için, cihazın maddeyi ihtiva etmesinin yararlılığına ilişkin onaylanmış kuruluş tarafından belirlenen verileri dikkate alır. Tıbbi ürünler danışma otoritesi, değişiklikler ile ilgili gerekli bütün dokümantasyonun alınmasından itibaren 60 gün içerisinde görüşünü bildirir. Tıbbi ürünler danışma otoritesi tarafından verilen bilimsel görüş olumsuz ise onaylanmış kuruluş, AB teknik dokümantasyon değerlendirme sertifikasına ek belge vermez. Onaylanmış kuruluş, tıbbi ürünler danışma otoritesine nihai kararını iletir. f) Tıbbi ürünler danışma otoritesinin, yardımcı madde hakkında, cihazın maddeyi ihtiva etmesiyle ilgili olarak önceden belirlenen risk veya fayda üzerinde bir etkiye sahip olabilecek bilgiler elde etmesi durumunda; tıbbi ürünler danışma otoritesi, bu bilgilerin, cihazın maddeyi ihtiva etmesiyle ilgili olarak önceden belirlenen risk veya fayda üzerinde bir etkisinin olup olmadığı konusunda onaylanmış kuruluşa tavsiyede bulunur. Onaylanmış kuruluş, uygunluk değerlendirme prosedürüyle ilgili değerlendirmesini yeniden incelerken bu tavsiyeyi dikkate alır. 5.3. Cansız veya cansız hale getirilmiş insan veya hayvan kaynaklı dokular veya hücreler ya da bunların türevleri kullanılarak imal edilen ya da bunları ihtiva eden cihazlar söz konusu olduğundaki prosedür 5.3.1. İnsan kaynaklı dokular veya hücreler ya da bunların türevleri a) 1 inci maddenin üçüncü fıkrasının (ç) bendi uyarınca bu Yönetmelik kapsamında olan, insan kaynaklı dokuların veya hücrelerin türevleri kullanılarak imal edilen cihazlar için ve cihazın fonksiyonuna yardımcı olan, İnsan Doku ve Hücreleri ile Bunlarla İlgili Merkezlerin Kalite ve Güvenliği Hakkında Yönetmeliğin kapsadığı insan kaynaklı dokuları veya hücreleri ya da bunların türevlerini bütünleşik bir parça olarak ihtiva eden cihazlar için, onaylanmış kuruluş, bir AB teknik dokümantasyon değerlendirme sertifikası düzenlemeden önce; insan kaynaklı dokuların veya hücrelerin ya da bunların türevlerinin, bağışlanması, tedarik edilmesi ve test edilmesi ile ilgili konular hakkındaki aynı Yönetmelik uyarınca Sağlık Bakanlığı ve/veya AB üyesi ülkeler tarafından belirlenen yetkili otoritelerin birinden (‘insan dokuları ve hücreleri yetkili otoritesi’) bilimsel görüş ister. Onaylanmış kuruluş, diğer hususların yanı sıra, söz konusu insan dokularının veya hücrelerinin cansız hale getirilmesi, bağışlanması, temin ve test edilmesine ve cihazın insan kaynaklı dokuları veya hücreleri ya da bunların türevlerini ihtiva etmesinin risk ve faydasına yönelik bilgiler sağlayan bir ön uygunluk değerlendirme özeti sunar. b) Gerekli bütün dokümantasyonun alınmasından itibaren 120 gün içerisinde, insan dokuları ve hücreleri yetkili otoritesi, görüşünü onaylanmış kuruluşa verir. c) İnsan dokuları ve hücreleri yetkili otoritesinin bilimsel görüşü ve bu görüşün olası güncellemeleri, onaylanmış kuruluşun cihaza ilişkin dokümantasyonuna dâhil edilir. Onaylanmış kuruluş, kararını verirken insan dokuları ve hücreleri yetkili otoritesinin bilimsel görüşünde ifade edilen görüşlere gereken önemi verir. Onaylanmış kuruluş, bilimsel görüş olumsuz ise sertifikayı vermez. İlgili insan dokuları ve hücreleri yetkili otoritesine nihai kararını iletir. ç) Cihazın ihtiva ettiği insan kaynaklı cansız dokular veya hücreler ya da bunların türevleriyle, özellikle de onların bağışlanması, test edilmesi veya temin edilmesiyle ilgili olarak herhangi bir değişiklik yapılmadan önce, imalatçı, amaçlanan değişiklikleri onaylanmış kuruluşa bildirir. Onaylanmış kuruluş, cihazın ihtiva ettiği insan kaynaklı dokuların veya hücrelerin ya da bunların türevlerinin kalitesinin ve güvenliliğinin sürdürüldüğünü teyit etmek için, ilk değerlendirmede yer alan otoriteye danışır. İlgili insan dokuları ve hücreleri yetkili otoritesi, bu değişiklerin, insan kaynaklı dokuların veya hücrelerin ya da bunların türevlerinin cihaza ihtivasının belirlenen fayda-risk oranı üzerinde olumsuz etkisinin olmadığından emin olmak amacıyla cihazın insan kaynaklı dokuları veya hücreleri ya da bunların türevlerini ihtiva etmesinin yararlılığına ilişkin onaylanmış kuruluş tarafından belirlenen verileri dikkate alır. Söz konusu otorite, amaçlanan değişiklere ilişkin gerekli bütün dokümantasyonun alınmasından itibaren 60 gün içerisinde görüşünü sunar. Onaylanmış kuruluş, bilimsel görüş olumsuz ise, AB teknik dokümantasyon sertifikasına ek belge vermez ve nihai kararını ilgili insan dokuları ve hücreleri yetkili otoritesine iletir. 5.3.2. Hayvan kaynaklı dokular veya hücreler ya da bunların türevleri Hayvan Kaynaklı Dokular Kullanılarak İmal Edilen Tıbbi Cihazlara Dair Yönetmelikte atıfta bulunulduğu şekilde, cansız hale getirilen hayvan dokuları kullanılarak ya da hayvan dokusundan elde edilen cansız ürünler kullanılarak imal edilen cihazlar söz konusu olduğunda, onaylanmış kuruluş, aynı Yönetmelikte belirtilen ilgili gereklilikleri uygular. 5.4. İnsan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazlar söz konusu olduğundaki prosedür a) Bir vücut açıklığı yoluyla insan vücuduna girmesi veya deriye uygulanması amaçlanan ve insan vücudu tarafından absorbe edilen ya da insan vücudunda lokal olarak dağılan maddelerden veya madde kombinasyonlarından oluşan cihazların kalitesi ve güvenliliği; uygulanabildiği hallerde ve yalnızca bu Yönetmeliğin kapsamadığı gerekliliklerle ilgili olarak, Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin Ek 1’inde absorbsiyonun, dağılımın, metabolizmanın, atılımın, lokal toleransın, toksisitenin, diğer cihazlar, tıbbi ürünler veya diğer maddelerle etkileşimin ve advers reaksiyonlar potansiyelinin değerlendirilmesi için belirtilen ilgili gereklilikler uyarınca doğrulanır. b) İlaveten, kullanım amaçlarını gerçekleştirmek amacıyla insan vücudu tarafından sistemik olarak absorbe edilen cihazlar ya da bu cihazların metabolizma ürünleri için, onaylanmış kuruluş; cihazın Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliğinin Ek 1’inde belirtilen ilgili gerekliliklere uygunluğu hakkında, aynı Yönetmelik uyarınca bu bent kapsamında hangisine danışıldığına bağlı olarak burada “tıbbi ürünler danışma otoritesi” adıyla atıfta bulunulacak olan Kurum ve/veya AB üyesi ülkeler tarafından atanan yetkili otoritelerin birinden ya da EMA’dan bilimsel görüş ister. c) Tıbbi ürünler danışma otoritesinin görüşü, gerekli bütün dokümantasyonun alınmasından itibaren 150 gün içerisinde hazırlanır. ç) Tıbbi ürünler danışma otoritesinin bilimsel görüşü ve bu görüşün olası güncellemeleri, onaylanmış kuruluşun cihaza ilişkin dokümantasyonuna dâhil edilir. Onaylanmış kuruluş, kararını verirken, bilimsel görüşte ifade edilen görüşlere gereken önemi verir ve nihai kararını tıbbi ürünler danışma otoritesine iletir. 6. Ayrı olarak kullanıldığında, 1 inci maddenin altıncı fıkrasında atıfta bulunulduğu şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün olduğu kabul edilebilen bir tıbbi maddeyi bütünleşik bir parça olarak ihtiva eden cihazlar söz konusu olduğunda parti doğrulaması Ayrı olarak kullanıldığında, 1 inci maddenin altıncı fıkrasının ikinci cümlesinde atıfta bulunulduğu şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün olduğu kabul edilebilen bir tıbbi maddeyi bütünleşik bir parça olarak ihtiva eden cihazların her bir partisinin imalatının tamamlanması sonrasında imalatçı; bu cihaz partisinin serbest bırakılmasını onaylanmış kuruluşa bildirir ve Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği uyarınca, ulusal bir laboratuvar veya Kurum tarafından bu amaçla kabul edilmiş bir laboratuvar tarafından düzenlenen, cihazda kullanılan insan kanı veya plazma türevi partisinin serbest bırakılması ile ilgili resmi sertifikayı onaylanmış kuruluşa gönderir.
III. BÖLÜM İDARİ HÜKÜMLER 7. İmalatçı veya yetkili temsilcisi; en son cihaz piyasaya arz edildikten sonra en az 10 yıl ve implante edilebilir cihazlar için en az 15 yıl süreyle, yetkili otoritelere sunmak üzere aşağıdakileri muhafaza eder: - AB uygunluk beyanını, - Bu Ekin 2.1 numaralı maddesinin beşinci bendinde atıfta bulunulan dokümantasyonu ve özellikle 2.2 numaralı maddesinin ikinci paragrafının (c) bendinde atıfta bulunulan prosedürlerden elde edilen verileri ve kayıtları, - Bu Ekin 2.4 numaralı maddesinde atıfta bulunulan değişikliklere ilişkin bilgileri, - Bu Ekin 4.2 numaralı maddesinde atıfta bulunulan dokümantasyonu ve - Bu Ekte atıfta bulunulduğu şekilde onaylanmış kuruluşun kararlarını ve raporlarını. 8. Kurum, imalatçının veya yetkili temsilcisinin, bu sürenin sona ermesinden önce iflas etmesi ya da ticari faaliyetini sonlandırması durumunda, bu Ekin 7 numaralı maddesinde atıfta bulunulan süre boyunca, atıfta bulunulan dokümantasyonun Kurumun tasarrufunda bulunmasını temin eder.
_______________________________________
Ek X TİP İNCELEMESİNE DAYALI UYGUNLUK DEĞERLENDİRMESİ 1. AB tip incelemesi; bir onaylanmış kuruluşun, cihazın teknik dokümantasyonu ve ilgili yaşam döngüsü süreçleri ve planlanan cihaz üretiminin ilgili temsili örneği de dâhil bir cihazın bu Yönetmeliğin ilgili hükümlerini yerine getirdiğini tespit ettiği ve belgelendirdiği prosedürdür. 2. Başvuru İmalatçı; değerlendirilmek üzere, bir onaylanmış kuruluşa başvuru yapar. Başvuru, aşağıdakileri içerir: - İmalatçının adı ile kayıtlı işyerinin adresini ve başvuru yetkili temsilci tarafından yapılıyorsa yetkili temsilcinin adı ile kayıtlı işyerinin adresini, - Ek II ve III’te atıfta bulunulan teknik dokümantasyonu. Başvuru sahibi, onaylanmış kuruluşa planlanan cihaz üretiminin temsili bir örneğini (tipi) sağlar. Onaylanmış kuruluş, gerektiğinde başka örnekler talep edebilir ve - Aynı tip için başka bir onaylanmış kuruluşa başvuru yapılmadığına dair yazılı bir beyanı ya da başka bir onaylanmış kuruluş tarafından reddedilmiş olan veya başka bir onaylanmış kuruluş nihai değerlendirmesini yapmadan önce imalatçı veya yetkili temsilcisi tarafından geri çekilmiş olan, aynı tipe yönelik önceki başvurular hakkında bilgileri. 3. Değerlendirme Onaylanmış kuruluş: a) İlgili teknolojiye ve klinik uygulamaya ilişkin kanıtlanmış bilgi ve deneyime sahip personel aracılığıyla başvuruyu inceler. Onaylanmış kuruluş, bu Yönetmeliğin ilgili gerekliliklerine uygunluğun değerlendirilmesine imkân sağlamak üzere, ilave testler yaptırarak ya da daha fazla kanıt isteyerek başvurunun tamamlanmasını talep edebilir. Onaylanmış kuruluş, cihazla ilgili yeterli fiziksel testleri veya laboratuvar testlerini yapar ya da bu tür testleri yapmasını imalatçıdan talep eder, b) Teknik dokümantasyonu, bu Yönetmeliğin cihaza uygulanabilir gerekliliklerine uygunluğuna yönelik inceler ve değerlendirir ve tipin bu dokümantasyona uygun olarak imal edildiğini doğrular; onaylanmış kuruluş ayrıca, parçaları 8 inci maddede atıfta bulunulan uygulanabilir standartlara veya ortak spesifikasyonlara uygun olarak tasarlanıp tasarlanmadıklarına göre kaydeder, c) Ek XIV’ün 4 numaralı maddesi uyarınca klinik değerlendirme raporunda imalatçı tarafından sunulan klinik kanıtları inceler. Onaylanmış kuruluş, yeterli klinik uzmanlığa sahip cihaz inceleyicileri istihdam eder ve gerektiği takdirde bu inceleme amaçları doğrultusunda, söz konusu cihazla ya da cihazın kullanıldığı klinik durumla ilgili doğrudan ve güncel deneyime sahip dış klinik uzmanlara başvurur, ç) Klinik kanıtların, değerlendirme kapsamındaki cihaza benzer veya eşdeğer olduğu iddia edilen cihazlardan alınan verilere kısmen veya tamamen dayandığı durumlarda, yeni endikasyonlar ve inovasyon gibi faktörleri dikkate alarak, bu tür verileri kullanmanın uygunluğunu değerlendirir. Onaylanmış kuruluş, iddia edilen eşdeğerlilik hakkındaki değerlendirmeleri ile uygunluğu göstermeye yönelik verilerin ilgililiği ve yeterliliği hakkındaki değerlendirmelerini açıkça dokümante eder, d) (ğ) bendinde atıfta bulunulan AB tip incelemesi raporunun bir bölümü olarak, klinik öncesi ve klinik değerlendirmenin değerlendirilmesi raporunda, değerlendirmesinin çıktısını açıkça dokümante eder, e) 8 inci maddede atıfta bulunulan standartların veya ortak spesifikasyonların uygulanmaması durumunda, imalatçı tarafından benimsenen çözümlerin bu Yönetmelikte belirlenen genel güvenlilik ve performans gerekliliklerini karşılayıp karşılamadığını doğrulamak üzere uygun değerlendirmeleri ve gerekli fiziksel testleri ya da laboratuvar testlerini yapar veya yaptırır. Cihazın amaçlandığı şekilde çalışması için başka bir cihaza veya cihazlara bağlanmasının gerekli olduğu durumlarda; imalatçı tarafından belirtilen karakteristiklere sahip olan bu tür cihaz veya cihazlara bağlandığında, cihazın genel güvenlilik ve performans gerekliliklerini karşıladığına dair kanıt sağlanır, f) İmalatçının ilgili uyumlaştırılmış standartları uygulamayı seçmesi durumunda, bu standartların gerçekten uygulanmış olup olmadığını doğrulamak için uygun değerlendirmeleri ve gerekli fiziksel testleri ya da laboratuvar testlerini yapar veya yaptırır; g) Gerekli değerlendirmelerin ve testlerin yapılacağı yer konusunda başvuru sahibi ile anlaşmaya varır ve ğ) (a) ila (f) bentleri kapsamında yapılan değerlendirmelerin ve testlerin sonuçları hakkında bir AB tip incelemesi raporu hazırlar. 4. Sertifika Tip bu Yönetmeliğe uyuyor ise onaylanmış kuruluş bir AB tip inceleme sertifikası düzenler. Sertifika; imalatçının adı ve adresini, tip incelemesi değerlendirmesinin sonuçlarını, sertifikanın geçerlilik şartlarını ve onaylanmış tipin tanımlanması için gereken verileri içerir. Sertifika, Ek XII uyarınca hazırlanır. Dokümantasyonun ilgili bölümleri sertifikaya eklenir ve bir sureti onaylanmış kuruluş tarafından muhafaza edilir. 5. Tipteki değişiklikler 5.1. Başvuru sahibi, onaylanmış tipe veya onun kullanım amacına ve kullanım şartlarına yönelik planlanan değişiklikler hakkında AB tip inceleme sertifikasını düzenleyen onaylanmış kuruluşu bilgilendirir. 5.2. Kullanım amacının ve kullanım şartlarının sınırlandırılması da dâhil onaylanmış cihazdaki değişiklikler; bu tür değişikliklerin genel güvenlilik ve performans gerekliliklerine veya ürünün kullanımına yönelik öngörülen şartlara uygunluğu etkileyebileceği durumlarda AB tip inceleme sertifikasını düzenleyen onaylanmış kuruluştan onay gerektirir. Onaylanmış kuruluş; planlanan değişiklikleri inceler, imalatçıya kararını bildirir ve AB tip incelemesi raporuna yapılan bir ek belge sunar. Onaylanmış tipteki her değişikliğin onayı, AB tip inceleme sertifikasına ek bir belge şeklinde olur. 5.3. Onaylanmış cihazın kullanım amacındaki ve kullanım şartlarındaki değişiklikler, kullanım amacının ve kullanım şartlarının sınırlandırılması hariç, yeni bir uygunluk değerlendirme başvurusunu gerekli kılar. 6. Spesifik ilave prosedürler AB teknik dokümantasyon değerlendirme sertifikasına yönelik herhangi bir atfın AB tip inceleme sertifikasına yönelik bir atıf olarak kabul edilmesi şartıyla, Ek IX’un 5 numaralı maddesi uygulanır. 7. İdari hükümler İmalatçı veya onun yetkili temsilcisi; en son cihaz piyasaya arz edildikten sonra en az 10 yıl ve implante edilebilir cihazlar için en az 15 yıl süreyle, yetkili otoritelere sunmak üzere aşağıdakileri muhafaza eder: - Bu Ekin 2 numaralı maddesinin üçüncü paragrafında atıfta bulunulan dokümantasyonu, - Bu Ekin 5 numaralı maddesinde atıfta bulunulan değişikliklere ilişkin bilgileri ve - AB tip inceleme sertifikalarının, bilimsel görüş ve raporların ve onların eklerinin/ek dokümanlarının suretlerini. Ek IX’un 8 numaralı maddesi uygulanır.
________________________________________________________
Ek XI ÜRÜN UYGUNLUK DOĞRULAMASINA DAYALI UYGUNLUK DEĞERLENDİRMESİ 1. Ürün uygunluk doğrulamasına dayalı uygunluk değerlendirmesinin hedefi; cihazların, bir AB tip inceleme sertifikasının düzenlenmiş olduğu tipe uygun olmasını ve onlara uygulanan bu Yönetmeliğin hükümlerini karşılamasını sağlamaktır. 2. Bir AB tip inceleme sertifikasının Ek X uyarınca düzenlenmiş olması durumunda, imalatçı; bu Ekin ya A Kısmında belirtilen prosedürü (üretim kalite güvencesi) ya da B Kısmında belirtilen prosedürü (ürün doğrulama) uygular 3. Bu Ekin 1 ve 2 numaralı maddelerine istisna olarak, Ek II ve Ek III’te belirtildiği şekilde hazırlanan teknik dokümantasyon ile birlikte bu Ekteki prosedürler sınıf IIa cihazların imalatçıları tarafından da uygulanabilir.
KISIM A ÜRETİM KALİTE GÜVENCESİ 4. İmalatçı; ilgili cihazların imalatı için onaylanan kalite yönetim sisteminin uygulanmasını sağlar, bu Ekin 6 numaralı maddesinde belirtildiği şekilde bir nihai doğrulama yapar ve 7 numaralı maddesinde atıfta bulunulan gözetime tabi olur. 5. İmalatçı; bu Ekin 4 numaralı maddesinde belirtilen yükümlülükleri yerine getirdiğinde, uygunluk değerlendirme prosedürünün kapsadığı cihaz için 19 uncu madde ve Ek IV uyarınca bir AB uygunluk beyanı hazırlar ve muhafaza eder. İmalatçının AB uygunluk beyanı düzenleyerek; ilgili cihazın, AB tip inceleme sertifikasında tanımlanan tipe uyduğunu ve bu Yönetmeliğin cihaza uygulanan gerekliliklerini karşıladığını garanti ve beyan ettiği kabul edilir. 6. Kalite yönetim sistemi 6.1. İmalatçı; kalite yönetim sisteminin değerlendirilmesi için bir onaylanmış kuruluşa başvuru yapar. Bu başvuru: - Ek IX’un 2.1 numaralı maddesinde listelenen tüm ögeleri, - Onaylanmış tipler için; Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyonu ve - Ek X’un 4 numaralı maddesinde atıfta bulunulan AB tip inceleme sertifikalarının bir suretini içerir. AB tip inceleme sertifikaları başvurunun yapıldığı onaylanmış kuruluş tarafından düzenlenmişse, teknik dokümantasyon ile güncellemelerine ve düzenlenen sertifikalara yönelik atıf başvuruya ayrıca dâhil edilir. 6.2. Kalite yönetim sisteminin uygulanması; AB tip inceleme sertifikasında tanımlanan tiple ve bu Yönetmeliğin cihazlara her aşamada uygulanan hükümleriyle uyumluluğu sağlayacak şekildedir. İmalatçı tarafından kalite yönetim sistemine yönelik benimsenen tüm ögeler, gereklilikler ve hükümler; kalite el kitabı ile kalite programları, kalite planları ve kalite kayıtları gibi yazılı politikalar ve prosedürler biçiminde, sistematik ve düzenli bir şekilde dokümante edilir. Bu dokümantasyon, özellikle, Ek IX’un 2.2 numaralı maddesinin (a), (b), (ç) ve (d) bentlerinde listelenen tüm ögelerin yeterli bir tanımını içerir. 6.3. Ek IX’un 2.3 numaralı maddesinin birinci ve ikinci paragrafı uygulanır. Kalite yönetim sistemi, cihazların AB tip inceleme sertifikasında tanımlanan tipe uymasını sağlamakta ve bu Yönetmeliğin ilgili hükümlerine uymakta ise onaylanmış kuruluş bir AB kalite güvence sertifikası düzenler. Onaylanmış kuruluş, sertifika düzenleme kararını imalatçıya bildirir. Bu karar, onaylanmış kuruluşun denetim sonuçlarını ve gerekçeli bir değerlendirmeyi içerir. 6.4. Ek IX’un 2.4 numaralı maddesi uygulanır. 7. Gözetim Ek IX’un; 3.1 numaralı maddesi, 3.2 numaralı maddesinin birinci, ikinci ve dördüncü bentleri, 3.3, 3.4, 3.6 ve 3.7 numaralı maddeleri uygulanır. Sınıf III cihazlar söz konusu olduğunda gözetim; üretilen veya satın alınan ham maddelerin veya kritik bileşenlerin miktarlarının tip için onaylandığına ve bitmiş cihazların miktarlarına karşılık geldiğine dair bir kontrolü de içerir. 8. Ayrı olarak kullanıldığında 1 inci maddenin altıncı fıkrasında atıfta bulunulduğu şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün olduğu kabul edilebilen bir tıbbi maddeyi bütünleşik bir parça olarak ihtiva eden cihazlar söz konusu olduğunda, parti doğrulaması Ayrı olarak kullanıldığında, 1 inci maddenin altıncı fıkrasının ikinci cümlesinde atıfta bulunulduğu şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün olduğu kabul edilebilen bir tıbbi maddeyi bütünleşik bir parça olarak ihtiva eden cihazların her bir partisinin imalatının tamamlanması sonrasında imalatçı; cihaz partisinin serbest bırakılmasını onaylanmış kuruluşa bildirir ve Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği uyarınca, ulusal bir laboratuvar veya Kurum tarafından bu amaçla kabul edilmiş bir laboratuvar tarafından düzenlenen, cihazda kullanılan insan kanı veya plazma türevi partisinin serbest bırakılması ile ilgili resmi sertifikayı onaylanmış kuruluşa gönderir. 9. İdari hükümler İmalatçı veya yetkili temsilcisi; en son cihaz piyasaya arz edildikten sonra en az 10 yıl ve implante edilebilir cihazlar için en az 15 yıl süreyle, yetkili otoritelere sunmak üzere aşağıdakileri muhafaza eder: - AB uygunluk beyanını, - Ek IX’un 2.1 numaralı maddesinin beşinci bendinde atıfta bulunulan dokümantasyonu, - Ek X’da atıfta bulunulan AB tip inceleme sertifikası dâhil, Ek IX’un 2.1 numaralı maddesinin sekizinci bendinde atıfta bulunulan dokümantasyonu, - Ek IX’un 2.4 numaralı maddesinde atıfta bulunulan değişikliklere ilişkin bilgileri ve - Ek IX’un 2.3, 3.3 ve 3.4 numaralı maddelerinde atıfta bulunulduğu şekilde onaylanmış kuruluşun kararlarını ve raporlarını. Ek IX’un 8 numaralı maddesi uygulanır. 10. Sınıf IIa cihazlara yönelik uygulama 10.1. Bu Ekin 5 numaralı maddesine istisna olarak, AB uygunluk beyanına binaen imalatçının; söz konusu sınıf IIa cihazların, Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona uygun olarak imal edildiğini ve onlara uygulanan bu Yönetmeliğin gerekliliklerini karşıladığını garanti ve beyan ettiği kabul edilir. 10.2. Sınıf IIa cihazlar söz konusu olduğunda onaylanmış kuruluş; bu Ekin 6.3 numaralı maddesinde atıfta bulunulan değerlendirmenin bir bölümü olarak, temsili bir temelde seçilen cihazlar için Ek II ve Ek III’te atıfta bulunulduğu şekilde teknik dokümantasyonun bu Yönetmelik ile uyumlu olup olmadığını değerlendirir. Onaylanmış kuruluş cihazların temsili bir örneğini veya örneklerini seçerken; teknolojinin yeniliğini, tasarımdaki benzerlikleri, teknolojiyi, imalat ve sterilizasyon yöntemlerini, kullanım amacını ve bu Yönetmelik uyarınca yürütülmüş olan fiziksel, kimyasal, biyolojik veya klinik özelliklere ilişkin olanlar gibi önceki ilgili değerlendirmelerin sonuçlarını dikkate alır. Onaylanmış kuruluş, alınan cihaz örneğine veya örneklerine yönelik gerekçesini dokümante eder. 10.3. Bu Ekin 10.2 numaralı maddesi kapsamındaki değerlendirmenin, söz konusu sınıf IIa cihazların Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona uyduğunu ve bu Yönetmeliğin onlara uygulanan gerekliliklerini karşıladığını teyit etmesi durumunda onaylanmış kuruluş; bu Ekin bu Kısmı uyarınca bir sertifika düzenler. 10.4. Cihazların ilk uygunluk değerlendirmesi için alınmış örneklere ilave olarak alınan örnekler, onaylanmış kuruluş tarafından, bu Ekin 7 numaralı maddesinde atıfta bulunulan gözetim değerlendirmesinin bir bölümü olarak değerlendirilir. 10.5. Bu Ekin 6 numaralı maddesine istisna olarak, imalatçı veya onun yetkili temsilcisi en son cihaz piyasaya arz edildikten sonra en az 10 yıl süreyle, yetkili otoritelere sunmak üzere aşağıdakileri muhafaza eder: - AB uygunluk beyanını, - Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyonu ve - Bu Ekin 10.3 numaralı maddesinde atıfta bulunulan sertifikayı. Ek IX’un 8 numaralı maddesi uygulanır.
KISIM B ÜRÜN DOĞRULAMASI 11. Ürün doğrulaması; imal edilmiş her cihazın incelenmesinden sonra imalatçının, 19 uncu madde ve Ek IV uyarınca bir AB uygunluk beyanı düzenlemek suretiyle, bu Ekin 14 ve 15 numaralı maddelerinde belirtilen prosedüre tabi olan cihazların AB tip-inceleme sertifikasında tanımlanan tipe uyduğunu ve bu Yönetmeliğin bu cihazlara uygulanan gerekliliklerini karşıladığını garanti ve beyan ettiğinin kabul edildiği prosedür olarak anlaşılır. 12. İmalatçı; imalat sürecinin, AB tip-inceleme sertifikasında tanımlanan tipe ve bu Yönetmeliğin uygulanan gerekliliklerine uygun cihazlar üretmesini sağlamak üzere gerekli tüm önlemleri alır. İmalatın başlamasından önce imalatçı; özellikle gerekli olduğunda sterilizasyonla ilgili olanlar başta olmak üzere imalat süreçlerini tanımlayan ve bununla birlikte homojen üretim ve uygun olduğu yerde cihazların AB tip-inceleme sertifikasında tanımlanan tipe ve onlara uygulanan bu Yönetmeliğin gerekliliklerine uygunluğunu sağlamak için uygulanacak, önceden belirlenmiş, rutin prosedürleri tanımlayan dokümanlar hazırlar. İlaveten, imalatçı; piyasaya steril olarak arz edilen cihazlar için ve yalnızca imalat sürecinin steriliteyi korumak ve sürdürmek için tasarlanan ilgili hususları için bu Ekin 6 ve 7 numaralı maddelerindeki hükümleri uygular. 13. İmalatçı; piyasaya arz sonrası klinik takip planı dâhil, piyasaya arz sonrası gözetim planı ile bu Yönetmeliğin Yedinci Kısım’ında belirtilen vijilans ve piyasaya arz sonrası gözetim ve denetim sistemine ilişkin hükümlerden doğan yükümlülüklerine uygunluğu sağlayan prosedürler oluşturmayı ve güncel tutmayı taahhüt eder. 14. Onaylanmış kuruluş, bu Ekin 15 numaralı maddesinde belirtildiği şekilde her ürünü inceleyerek ve test ederek cihazın bu Yönetmeliğin gerekliliklerine uygunluğunu doğrulamak amacıyla uygun incelemeler ve testler yürütür. Birinci paragrafta atıfta bulunulan incelemeler ve testler, imalat sürecinin steriliteyi korumak için tasarlanan ilgili hususlarına uygulanmaz. 15. Her ürünün incelenmesi ve test edilmesi yoluyla doğrulama 15.1. Her cihaz ayrı ayrı incelenir ve uygun olduğu hallerde cihazların AB tip-inceleme sertifikasında tanımlanan tipe ve bu Yönetmeliğin onlara uygulanan gerekliliklerine uygunluğunu doğrulamak amacıyla, 8 inci maddede atıfta bulunulan ilgili standart veya standartlarda tanımlandığı şekilde uygun fiziksel testler veya laboratuvar testleri ya da eşdeğer testler ve değerlendirmeler yürütülür. 15.2. Onaylanmış kuruluş; onaylanmış her cihaza, kendi kimlik numarasını iliştirir ya da iliştirtir ve yapılan testler ve değerlendirmeler ile ilgili bir AB ürün doğrulama sertifikası hazırlar. 16. Ayrı olarak kullanıldığında 1 inci maddenin altıncı fıkrasında atıfta bulunulan insan kanı veya insan plazmasından elde edilen bir tıbbi ürün olduğu kabul edilebilen bir tıbbi maddeyi bütünleşik bir parça olarak ihtiva eden cihazlar söz konusu olduğunda, parti doğrulaması Ayrı olarak kullanıldığında, 1 inci maddenin altıncı fıkrasının ikinci cümlesinde atıfta bulunulduğu şekilde insan kanı veya insan plazmasından elde edilen bir tıbbi ürün olduğu kabul edilebilen bir tıbbi maddeyi, bütünleşik bir parça olarak ihtiva eden cihazların her bir partisinin imalatının tamamlanması sonrasında imalatçı; cihaz partisinin serbest bırakılmasını onaylanmış kuruluşa bildirir ve Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği uyarınca, ulusal bir laboratuvar veya Kurum tarafından bu amaçla kabul edilmiş bir laboratuvar tarafından düzenlenen, cihazda kullanılan insan kanı veya plazma türevi partisinin serbest bırakılması ile ilgili resmi sertifikayı onaylanmış kuruluşa gönderir. 17. İdari hükümler İmalatçı veya onun yetkili temsilcisi; en son cihaz piyasaya arz edildikten sonra en az 10 yıl ve implante edilebilir cihazlar için 15 yıl süreyle, yetkili otoritelere sunmak üzere aşağıdakileri muhafaza eder: - AB uygunluk beyanını, - Bu Ekin 12 numaralı maddesinde atıfta bulunulan dokümantasyonu, - Bu Ekin 15.2 numaralı maddesinde atıfta bulunulan sertifikayı ve - Ek X’da atıfta bulunulan AB tip-inceleme sertifikasını. Ek IX’un 8 numaralı maddesi uygulanır. 18. Sınıf IIa cihazlara yönelik uygulama 18.1 Bu Ekin 11 numaralı maddesine istisna olarak, AB uygunluk beyanına binaen imalatçının; söz konusu sınıf IIa cihazların, Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona uygun olarak imal edilmesini ve bu Yönetmeliğin bu cihazlara uygulanan gerekliliklerini karşılamasını garanti ve beyan ettiği kabul edilir. 18.2. Bu Ekin 14 numaralı maddesi uyarınca onaylanmış kuruluş tarafından yürütülen doğrulamanın; söz konusu sınıf IIa cihazların, Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona ve bu Yönetmeliğin bu cihazlara uygulanan gerekliliklerine uygunluğunu teyit etmesi amaçlanır 18.3. Bu Ekin 18.2 numaralı maddesinde atıfta bulunulan doğrulama, söz konusu sınıf IIa cihazların, Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyona ve bu Yönetmeliğin bu cihazlara uygulanan gerekliliklerine uygun olduğunu teyit ederse, onaylanmış kuruluş bu Ekin bu Kısmı uyarınca bir sertifika düzenler. 18.4. Bu Ekin 17 numaralı maddesine istisna olarak, imalatçı veya yetkili temsilcisi, en son cihaz piyasaya arz edildikten sonra en az 10 yıl süreyle yetkili otoritelere sunmak üzere aşağıdakileri muhafaza eder: - AB uygunluk beyanını, - Ek II ve Ek III’te atıfta bulunulan teknik dokümantasyonu ve - Bu Ekin 18.3 numaralı maddesinde atıfta bulunulan sertifikayı. Ek IX’un 8 numaralı maddesi uygulanır.
____________________________________________________
Ek XII ONAYLANMIŞ KURULUŞ TARAFINDAN DÜZENLENEN SERTİFİKALAR
I. BÖLÜM GENEL GEREKLİLİKLER 1. Sertifikalar, Türkçe ve/veya AB resmi dillerinden birinde düzenlenir. 2. Her sertifika, yalnızca bir uygunluk değerlendirme prosedürüne atıfta bulunur. 3. Sertifikalar, yalnızca bir imalatçı için düzenlenir. Sertifikada yer alan imalatçının adı ve adresi, 30 uncu maddede atıfta bulunulan elektronik sisteme kaydedilenler ile aynı olur. 4. Sertifikaların kapsamı, kapsadığı cihazı veya cihazları açık bir şekilde tanımlar: a) AB teknik dokümantasyon değerlendirme sertifikaları, AB tip inceleme sertifikaları ve AB ürün doğrulama sertifikaları; ad, model ve tip dâhil olmak üzere cihaz veya cihazların açık bir tanımlamasını, imalatçı tarafından kullanım talimatına eklendiği şekilde ve cihazın tabi tutulduğu uygunluk değerlendirme prosedürüyle ilgili kullanım amacını, risk sınıfını ve 27 nci maddenin altıncı fıkrasında atıfta bulunulduğu şekilde Temel UDI-DI’yı içerir. b) AB kalite yönetim sistemi sertifikaları ve AB kalite güvence sertifikaları; cihazların veya cihaz gruplarının tanımlamasını, risk sınıfını ve sınıf IIb cihazlar için kullanım amacını içerir. 5. Onaylanmış kuruluş; talep üzerine, hangi (münferit) cihazların sertifika kapsamında olduğunu gösterebilecektir. Onaylanmış kuruluş; sertifika kapsamındaki cihazların sınıflandırmaları dâhil belirlenmesini sağlayan bir sistem kurar. 6. Sertifikalar; kapsadığı cihaz/cihazların piyasaya arzı için bu Yönetmelik uyarınca düzenlenmiş başka bir sertifikanın gerekmesi durumunda buna ilişkin bir not içerir. 7. 52 nci maddenin altıncı fıkrası uyarınca bir onaylanmış kuruluşun dâhil olmasını gerektiren sınıf I cihazlara yönelik AB kalite yönetim sistemi sertifikaları ve AB kalite güvence sertifikaları; onaylanmış kuruluş tarafından yapılan kalite yönetim sistemi denetiminin, söz konusu fıkra kapsamında gerekli olan hususlarla sınırlandırıldığına dair bir ifade içerir. 8. Bir sertifikanın ilave yapılmış, tadil edilmiş veya yeniden düzenlenmiş olması durumunda yeni sertifika; değişikliklerin tanımı ile birlikte, önceki sertifikaya ve onun düzenlenme tarihine bir atıf içerir.
II. BÖLÜM SERTİFİKALARIN ASGARİ İÇERİĞİ 1. Onaylanmış kuruluşun adı, adresi ve kimlik numarası, 2. İmalatçının ve uygulanabildiği hallerde yetkili temsilcinin adı ve adresi, 3. Sertifikayı tanımlayan özgün numara, 4. Hâlihazırda verilmiş ise 31 inci maddenin ikinci fıkrasında atıfta bulunulan, imalatçının münferit kayıt numarası, 5. Düzenlenme tarihi, 6. Geçerlilik (bitiş) tarihi, 7. Uygulanabildiği hallerde; bu Ekin I. Bölümünün 4 numaralı maddesinde belirtildiği şekilde cihazın veya cihazların açıkça tanımlanması için gerekli veriler, 8. Uygulanabildiği hallerde; bu Ekin I. Bölümünün 8 numaralı maddesinde belirtildiği şekilde önceki her sertifikaya atıf, 9. Bu Yönetmeliğe ve uygunluk değerlendirmesinin yürütüldüğü ilgili Eke atıf, 10. Gerçekleştirilen incelemeler ve testler, örneğin; ilgili ortak spesifikasyonlara, uyumlaştırılmış standartlara, test raporlarına ve denetim raporuna/raporlarına atıf, 11. Uygulanabildiği hallerde; teknik dokümantasyonun ilgili bölümlerine veya kapsadığı cihaz/cihazların piyasaya arzı için gerekli olan diğer sertifikalara atıf, 12. Uygulanabildiği hallerde; onaylanmış kuruluş tarafından yapılan gözetim hakkında bilgiler, 13. Onaylanmış kuruluşun ilgili Eke ilişkin uygunluk değerlendirmesinin sonuçları, 14. Sertifikanın geçerliliğine yönelik koşullar veya sınırlamalar, 15. İlgili mevzuat uyarınca onaylanmış kuruluşun yasal olarak geçerli imzası.
_______________________________
Ek XIII ISMARLAMA İMAL EDİLEN CİHAZLARA YÖNELİK PROSEDÜR 1. Ismarlama imal edilen cihazlar için, imalatçı veya yetkili temsilcisi aşağıdaki bilgilerin tümünü içeren bir beyan hazırlar: - İmalatçının ve tüm imalat tesislerinin adı ve adresi, - Uygulanabildiği hallerde; yetkili temsilcinin adı ve adresi, - Söz konusu cihazın tanımlanmasına olanak sağlayan veriler, - Cihazın yalnızca; ad, kısaltma veya nümerik bir kod ile tanımlanmış belirli bir hasta veya kullanıcı tarafından kullanılmasının amaçlandığına dair bir ifade, - Mesleki yeterliliklerine binaen yetkilendirilen, reçeteyi yazan kişinin adı ve uygulanabildiği yerde, ilgili sağlık kuruluşunun adı, - Reçetede belirtildiği şekilde ürünün spesifik karakteristikleri, - Söz konusu cihazın Ek I’de belirtilen genel güvenlilik ve performans gerekliliklerine uyduğuna dair ve uygulanabildiği hallerde gerekçeleri ile birlikte hangi genel güvenlilik ve performans gerekliliklerinin tam olarak karşılanmadığını belirten bir açıklama, - Uygulanabildiği hallerde; cihazın, bir insan kanı veya plazma türevi veya insan kaynaklı ya da Hayvan Kaynaklı Dokular Kullanılarak İmal Edilen Tıbbi Cihazlara Dair Yönetmelikte atıfta bulunulduğu şekilde hayvan kaynaklı dokular veya hücreler dâhil bir tıbbi madde içerdiğine veya ihtiva ettiğine dair bir işaret. 2. İmalatçı; bu Yönetmeliğin gerekliliklerine uygunluğun değerlendirilmesine imkân sağlayacak şekilde, imalat tesisini veya tesislerini belirten ve beklenen performans dâhil cihazın tasarım, imalat ve performansını anlamaya olanak sağlayan dokümantasyonu, Kurum için hazır bulundurmayı taahhüt eder. 3. İmalatçı; imalat sürecinin, bu Ekin 2 numaralı maddesinde atıfta bulunulan dokümantasyona uygun cihazlar imal edilmesini sağlaması amacıyla gerekli tüm önlemleri alır. 4. Bu Ekin 1 numaralı maddesinde belirtilen bildirim; cihazın piyasaya arz edilmesinden sonra en az 10 yıllık bir süreyle muhafaza edilir. İmplante edilebilir cihazlar söz konusu olduğunda bu süre, en az 15 yıl olur. Ek IX’un 8 numaralı maddesi uygulanır. 5. İmalatçı; Ek XIV’ün B Kısmında atıfta bulunulduğu şekilde piyasaya arz sonrası klinik takipten elde edilenler de dâhil, üretim sonrası aşamada kazanılan deneyimi inceler ve dokümante eder; gerekli herhangi bir düzeltici faaliyet uygulamak için uygun yöntemleri kullanır. Bu bağlamda, olumsuz olayları veya saha güvenliği düzeltici faaliyetlerini veya her ikisini, 85 inci maddenin birinci fıkrası uyarınca yetkili otoritelere, haberdar olur olmaz raporlar.
______________________________________________
Ek XIV KLİNİK DEĞERLENDİRME VE PİYASAYA ARZ SONRASI KLİNİK TAKİP
KISIM A KLİNİK DEĞERLENDİRME 1. İmalatçılar; bir klinik değerlendirmeyi planlamak, sürekli olarak gerçekleştirmek ve dokümante etmek için: a) Asgari olarak, aşağıdakileri içeren bir klinik değerlendirme planı oluşturur ve günceller: - İlgili klinik verilerle desteklenmesi gereken genel güvenlilik ve performans gerekliliklerinin tanımlamasını, - Cihazın kullanım amacının açıklamasını, - Belirgin endikasyonlar ve kontrendikasyonlar ile birlikte hedeflenen gruplara yönelik net açıklamayı, - İlgili ve belirlenmiş klinik çıktı parametreleri ile birlikte, hastalara yönelik amaçlanan klinik faydaların ayrıntılı açıklamasını, - Artık risklerin ve yan etkilerin belirlenmesine açık bir referansla, klinik güvenliğin nitel ve nicel hususlarının incelenmesi için kullanılacak yöntemlerin açıklamasını, - Tıptaki en son gelişmeler ışığında cihazın çeşitli endikasyonlarına ve kullanım amacı veya amaçlarına yönelik fayda-risk oranının kabul edilebilirliğini belirlemek için kullanılacak parametrelerin açıklamasını ve listesini, - Farmasötiklerin, cansız hayvan veya insan dokularının kullanımı gibi, spesifik bileşenlerle ilgili fayda-risk konularının nasıl ele alınacağının göstergesini ve - İnsan üzerindeki ilk çalışmalar, fizibilite ve pilot çalışmalar gibi keşif amaçlı araştırmalardan, asıl klinik araştırmalar gibi teyit edici araştırmalara kadar ilerlemeyi gösteren klinik gelişim planını ve kilit noktaların göstergesi ve potansiyel kabul kriterlerinin açıklaması ile birlikte bu Ekin Kısım B’sinde atıfta bulunulduğu şekilde bir PMCF. b) Cihaza ve kullanım amacına ilişkin mevcut klinik verileri ve klinik kanıtlardaki boşlukları sistematik bilimsel literatür taraması yoluyla tanımlar, c) Cihazın güvenlilik ve performansını tesis etmek için uygunluklarını değerlendirme yoluyla, ilgili tüm klinik verileri değerlendirir, ç) Klinik gelişim planına uygun olarak, doğru şekilde tasarlanmış klinik araştırmalar vasıtasıyla, çözümlenmemiş hususları ele almak üzere gerekli olan yeni veya ilave klinik verileri oluşturur, d) Cihazın klinik faydaları dâhil, güvenliliğine ve klinik performansına ilişkin sonuçlara ulaşmak amacıyla, ilgili tüm klinik verileri analiz eder. 2. Klinik değerlendirme; ayrıntılı ve nesnel olur ve hem olumlu hem de olumsuz verileri dikkate alır. Klinik değerlendirmenin derinliği ve kapsamı; imalatçının cihaza ilişkin iddialarıyla birlikte, söz konusu cihazın niteliği, sınıflandırması, kullanım amacı ve riskleriyle orantılı ve bunlara uygun olur. 3. Klinik değerlendirme, söz konusu cihaza eşdeğerliliği gösterilebilen bir cihazla ilgili klinik verilere dayanabilir. Aşağıdaki teknik, biyolojik ve klinik karakteristikler eşdeğerliliğin gösterimi için göz önünde bulundurulur: - Teknik: Cihaz, benzer tasarıma sahip olup benzer kullanım şartları altında kullanılır. Enerji yoğunluğu, çekme dayanımı, viskozite, yüzey karakteristikleri, dalga boyu gibi fiziko-kimyasal özellikler ve yazılım algoritması dâhil benzer spesifikasyonlara ve özelliklere sahiptir. İlgili olduğu yerde, benzer kurulum yöntemleri kullanır ve benzer çalışma prensiplerine ve kritik performans gerekliliklerine sahiptir. - Biyolojik: Cihaz, aynı insan dokuları veya vücut sıvılarıyla temas halinde benzer temas türü ve süresi ve bozunma ürünleri ve ayrıştırılabilenler de dâhil maddelerin benzer salınım karakteristikleri için aynı materyalleri veya maddeleri kullanır; - Klinik: Cihaz, vücudun aynı bölgesinde, yaş, anatomi ve fizyoloji bakımından olanlar da dâhil benzer bir popülasyonda, benzer hastalık şiddeti ve evresi dâhil olmak üzere aynı klinik durum veya amaç için kullanılır. Aynı kullanıcı türüne sahiptir ve belirli bir kullanım amacına yönelik beklenen klinik etki açısından ilgili benzer kritik performansa sahiptir. Birinci paragrafta listelenen karakteristikler, cihazın güvenlilik ve klinik performansında klinik olarak önemli farklılıklar oluşturmayacak ölçüde benzer olur. Eşdeğerlilik değerlendirmeleri, uygun bilimsel gerekçelere dayanır. İmalatçıların, eşdeğerlilik iddialarını gerekçelendirmek amacıyla, eşdeğerlilik iddia ettikleri cihazlarla ilgili verilere yeterli seviyede erişim sağladıkları açık bir şekilde gösterilir. 4. Klinik değerlendirme sonuçları ve dayandırıldığı klinik kanıtlar, cihazın uygunluk değerlendirmesini destekleyen bir klinik değerlendirme raporunda dokümante edilir. Klinik olmayan test yöntemlerinden elde edilen klinik olmayan veriler ile birlikte klinik kanıtlar ve diğer ilgili dokümantasyon; imalatçının, genel güvenlilik ve performans gerekliliklerine uygunluğu göstermesine imkân verir ve söz konusu cihaza ilişkin teknik dokümantasyonun bir bölümü olur. Klinik değerlendirmede dikkate alınan olumlu ve olumsuz verilerin her ikisi birlikte teknik dokümantasyona dâhil edilir.
KISIM B PİYASAYA ARZ SONRASI KLİNİK TAKİP 5. Piyasaya arz sonrası klinik takip, 61 inci maddede ve bu Ekin A Kısmında atıfta bulunulan klinik değerlendirmeyi güncelleyen devamlı bir süreç olarak anlaşılır ve imalatçının piyasaya arz sonrası gözetim planında ele alınır. Piyasaya arz sonrası klinik takibi yürütürken imalatçı; cihazın beklenen kullanım ömrü süresince güvenliliğini ve performansını teyit etmek, tanımlanmış risklerin sürekli kabul edilebilirliğini sağlamak ve gerçek kanıtlara dayanarak yeni ortaya çıkan riskleri tespit etmek amacıyla, CE işareti taşıyan ve ilgili uygunluk değerlendirme prosedüründe belirtildiği şekilde kullanım amacı dâhilinde piyasaya arz edilen veya hizmete sunulan bir cihazın insan vücudu üzerinde veya içerisinde kullanımından elde edilen klinik verileri proaktif olarak toplar ve değerlendirir. 6. Piyasaya arz sonrası klinik takip; piyasaya arz sonrası klinik takip planında belirtilen dokümante edilmiş yöntem uyarınca gerçekleştirilir. 6.1. Piyasaya arz sonrası klinik takip planı, aşağıdaki amaçlarla, klinik verileri proaktif olarak toplamak ve değerlendirmek için yöntemler ve prosedürler belirtir: a) Öngörülen kullanım ömrü boyunca cihazın güvenlilik ve performansını teyit etmek. b) Önceden bilinmeyen yan etkileri tanımlamak ve tanımlanmış yan etkileri ve kontrendikasyonları izlemek. c) Gerçek kanıtlara dayanarak, yeni ortaya çıkan riskleri tanımlamak ve analiz etmek. ç) Ek I’in 1 ve 9 numaralı maddelerinde atıfta bulunulan fayda-risk oranının sürekli kabul edilebilirliğini sağlamak. d) Kullanım amacının uygunluğunu teyit etmek amacıyla, cihazın olası sistematik yanlış kullanımını veya endikasyon dışı kullanımını tanımlamak. 6.2. PMCF planı, asgari olarak aşağıdakileri içerir: a) Kazanılan klinik tecrübenin toplanması, kullanıcılardan geri bildirim alınması, bilimsel literatürün ve diğer klinik veri kaynaklarının taranması gibi, uygulanacak PMCF’nin genel yöntemlerini ve prosedürlerini. b) Uygun kayıtların veya PMCF çalışmalarının değerlendirilmesi gibi, uygulanacak PMCF’nin spesifik yöntemlerini ve prosedürlerini. c) (a) ve (b) bentlerinde atıfta bulunulan yöntemlerin ve prosedürlerin uygunluğuna yönelik gerekçeyi. ç) Bu Ekin 4 numaralı maddesinde atıfta bulunulan klinik değerlendirme raporunun ilgili bölümlerine ve Ek I’in 3 numaralı maddesinde atıfta bulunulan risk yönetimine atfı. d) PMCF’nin kapsayacağı spesifik hedefleri. e) Eşdeğer veya benzer cihazlarla ilgili klinik verilerin değerlendirmesini. f) İlgili ortak spesifikasyonlara, imalatçı tarafından kullanılmış ise uyumlaştırılmış standartlara ve PMCF’ye yönelik ilgili kılavuza atfı. g) İmalatçı tarafından üstlenilecek, PMCF verilerinin analizi ve raporlaması gibi, PMCF faaliyetlerine yönelik detaylandırılmış ve yeterli bir şekilde gerekçelendirilmiş zaman çizelgesini. 7. İmalatçı, PMCF bulgularını analiz eder; sonuçları, klinik değerlendirme raporunun ve teknik dokümantasyonun bir bölümü olan PMCF değerlendirme raporunda dokümante eder. 8. PMCF değerlendirme raporunun sonuçları, 61 inci madde ve bu Ekin A Kısmında atıfta bulunulan klinik değerlendirmede ve Ek I’in 3 numaralı maddesinde atıfta bulunulan risk yönetiminde dikkate alınır. PMCF vasıtasıyla, önleyici ve/veya düzeltici önlemlere yönelik ihtiyaç tanımlandıysa, imalatçı bunları uygular.
_________________________________________________________
Ek XV KLİNİK ARAŞTIRMALAR
I. BÖLÜM GENEL GEREKLİLİKLER 1. Etik ilkeler Klinik araştırmanın; çalışmaya yönelik ihtiyacın ve gerekçenin ön değerlendirmesinden sonuçların yayımlanmasına kadar her adımı, kabul görmüş etik ilkelere uygun olarak yürütülür. 2. Yöntemler 2.1. Klinik araştırmalar, en son bilimsel ve teknik bilgileri yansıtan ve 62 nci maddenin birinci fıkrasında atıfta bulunulduğu şekilde cihazların güvenliliğine, performansına ve fayda-riskleri ile ilgili hususlara ilişkin imalatçının iddialarını teyit edecek veya çürütecek şekilde tanımlanmış uygun bir araştırma planına dayanılarak gerçekleştirilir. Klinik araştırmalar, sonuçların bilimsel geçerliliğini garanti etmek üzere yeterli sayıda gözlemi içerir. Tasarıma ve seçilmiş istatistiksel metodolojiye yönelik gerekçe, bu Ekin II. Bölümünün 3.6 numaralı maddesinde açıklandığı şekilde sunulur. 2.2. Klinik araştırmayı gerçekleştirmek için kullanılan prosedürler, araştırma kapsamındaki cihaza uygun olur. 2.3. Klinik araştırmayı gerçekleştirmek için kullanılan araştırma metodolojileri, araştırma kapsamındaki cihaza uygun olur. 2.4. Klinik araştırmalar; klinik araştırma planı uyarınca, hedef hasta popülasyonunda cihazın normal kullanım koşullarını temsil eden bir klinik ortamda ve yeterli sayıda hedef kullanıcı ile gerçekleştirilir. Klinik araştırmalar, Ek XIV’ün A Kısmında atıfta bulunulduğu şekilde klinik değerlendirme planı doğrultusunda yapılır. 2.5. Cihazın, özellikle güvenliliğini ve performansını ilgilendiren tüm uygun teknik ve işlevsel özellikleri ve bunların beklenen klinik çıktıları, araştırmanın tasarımında uygun bir şekilde ele alınır. Cihazın teknik ve işlevsel özelliklerinin ve beklenen ilgili klinik çıktılarının bir listesi temin edilir. 2.6. Klinik araştırmanın sonlanım noktaları; cihazın kullanım amacını, klinik faydalarını, performansını ve güvenliliğini ele alır. Sonlanım noktaları, bilimsel olarak geçerli metodolojiler kullanılarak belirlenir ve değerlendirilir. Birincil sonlanım noktası, cihaza uygun ve klinik olarak ilgili olur. 2.7. Araştırmacılar, cihazla ilgili teknik ve klinik verilere erişime sahip olur. Bir araştırmanın yürütülmesine dâhil olan personel; araştırma amaçlı cihazın doğru kullanımı, klinik araştırma planı ve iyi klinik uygulamaları konusunda yeterli bir şekilde bilgilendirilir ve eğitilir. Bu eğitim; gerektiğinde sponsor tarafından düzenlenir, doğrulanır ve uygun bir şekilde dokümante edilir. 2.8. Araştırmacı tarafından imzalanan klinik araştırma raporu, klinik araştırma süresince toplanan tüm verilerin eleştirel bir değerlendirmesini kapsar ve herhangi bir olumsuz bulguyu da içerir.
II. BÖLÜM KLİNİK ARAŞTIRMAYA YÖNELİK BAŞVURUYA İLİŞKİN DOKÜMANTASYON 62 nci maddenin kapsadığı araştırma amaçlı cihazlar için sponsor, aşağıdaki dokümanlarla birlikte 70 inci madde uyarınca bir başvuru hazırlar ve sunar: 1. Başvuru formu Başvuru formu, aşağıdakilere ilişkin bilgileri içerecek şekilde, usulünce doldurulur: 1.1. Sponsorun adı, adresi ve iletişim bilgileri ve mevcutsa 62 nci maddenin ikinci fıkrası uyarınca yurt içinde yerleşik olan yasal temsilcisinin adı, adresi ve iletişim bilgileri, 1.2. 1.1’dekilerden farklı ise, klinik araştırma amaçlı cihazın imalatçısının ve mevcutsa yetkili temsilcisinin adı, adresi ve iletişim bilgileri, 1.3. Klinik araştırmanın başlığı, 1.4. Klinik araştırma başvurusunun durumu (örn, ilk başvuru, yeniden başvuru, önemli değişiklik), 1.5. Klinik değerlendirme planı hakkında ayrıntılar ve/veya ilgili plana atıf, 1.6. Başvuru, hâlihazırda başvurusu yapılmış bir cihaza ilişkin bir yeniden başvuru ise önceki başvurunun tarih/tarihleri ve referans numarası/ numaraları ya da önemli değişiklik durumunda, ilk başvuruya atıf. Sponsor, yetkili otoritenin veya etik kurulun önceki gözden geçirme bulgularını ele almak üzere yapılan değişiklikler başta olmak üzere önceki başvurudan itibaren ortaya çıkan değişikliklerin tamamını, bu değişikliklere yönelik bir gerekçe ile birlikte belirtir. 1.7. Başvuru, ilaç ve biyolojik ürünlerin klinik araştırmalarına yönelik başvuruya paralel olarak sunuluyorsa, bu klinik çalışmanın resmî kayıt numarasına atıf, 1.8. Klinik araştırmanın, çok merkezli veya çok uluslu çalışmanın bir parçası olarak yürütüleceği ülkelerin başvuru sırasında tanımlanması, 1.9. Araştırma amaçlı cihazın kısa bir tanımlaması, sınıflandırması, cihazın ve cihaz tipinin tanımlanması için gerekli diğer bilgiler, 1.10. Cihazın, bir insan kanı veya plazma türevi dâhil bir tıbbi madde içerip içermediğine ya da insan veya hayvan kaynaklı cansız dokular veya hücreler ya da bunların türevleri kullanılarak imal edilip edilmediğine ilişkin bilgiler, 1.11. Klinik araştırmanın amacı veya amaçları, gönüllülerin sayısı ve cinsiyeti, gönüllü seçimine yönelik kriterler, 18 yaş altında gönüllülerin olup olmadığı, araştırmanın tasarımı (örn. kontrollü ve/veya randomize çalışmalar), klinik araştırmanın planlanan başlama ve sonlandırma tarihleri dâhil olmak üzere klinik araştırma planının özeti, 1.12. Mevcutsa, karşılaştırma cihazına ilişkin bilgiler, sınıflandırması ve karşılaştırma cihazının tanımlanması için gerekli diğer bilgiler, 1.13. Klinik araştırmacının ve araştırma merkezinin, klinik araştırma planı uyarınca klinik araştırmayı yürütme kabiliyetine ilişkin sponsorun kanıtları, 1.14. Araştırmanın öngörülen başlama tarihi ve süresi ile ilgili ayrıntılar, 1.15. Hâlihazırda bir onaylanmış kuruluşla çalışılıyor iken bir klinik araştırma başvurusu yapılıyor ise başvuru aşamasında bu kuruluşu tanımlayıcı ayrıntılar, 1.16. Yetkili otoritenin; başvuruyu değerlendirmiş ya da değerlendirmekte olan etik kurul ile temasa geçebileceğinden, sponsorun haberdar olduğunun teyidi ve 1.17. Bu Ekin 4.1 numaralı maddesinde atıfta bulunulan beyanname. 2. Araştırmacı Broşürü Araştırmacı broşürü, araştırma amaçlı cihaza ilişkin araştırmayla alakalı ve başvuru anında mevcut olan klinik ve klinik dışı bilgileri içerir. Araştırmacı broşürüne yönelik güncellemeler veya yeni ortaya çıkan ilgili diğer bilgiler, araştırmacının dikkatine zamanında sunulur. Araştırmacı broşürü, açıkça tanımlanır ve özellikle aşağıdakileri içerir: 2.1. Kullanım amacına, risk sınıflandırmasına ve Ek VIII uyarınca uygulanabilir sınıflandırma kuralına, cihazın tasarımına ve imalatına ilişkin bilgiler dâhil cihazın tanımlaması ve açıklaması ile cihazın önceki ve benzer nesillerine atfı, 2.2. Depolama ve kullanım gereklilikleri dâhil, kuruluma, bakıma, hijyen standartlarını korumaya ve kullanıma yönelik imalatçının talimatları ile birlikte mevcut olduğu ölçüde, piyasaya arz edildiğinde etiket üzerine yerleştirilecek bilgileri ve cihazla birlikte temin edilecek kullanım talimatını. Buna ilave olarak, gerek duyulan ilgili her türlü eğitime ilişkin bilgileri, 2.3. Uygulanabilir olduğunda, tasarım içi hesaplamalar, in vitro testler, eks vivo testler, hayvan testleri, mekanik veya elektriksel testler, güvenilirlik testleri, sterilizasyon validasyonu, yazılım doğrulaması ve validasyonu, performans testleri, biyouyumluluk ve biyolojik güvenlik değerlendirmesi başta olmak üzere ilgili klinik öncesi testlere ve deneysel verilere dayalı klinik öncesi değerlendirmeyi, 2.4. Mevcut klinik verileri, özellikle: - cihazın ve/veya eşdeğer ya da benzer cihazların güvenliliğine, performansına, hastalara yönelik klinik faydalarına, tasarım karakteristiklerine ve kullanım amacına ilişkin mevcut ilgili bilimsel literatürden elde edilen klinik verileri; - aynı imalatçının eşdeğer ya da benzer cihazların güvenliliğine, performansına, hastalara yönelik klinik faydalarına, tasarım karakteristiklerine ve kullanım amacına ilişkin, piyasada bulunma süresi ve performans, klinik fayda ve güvenlilikle ilgili konuların değerlendirmesi ve alınan düzeltici faaliyetler dâhil, mevcut diğer ilgili klinik verileri. 2.5. Bilinen veya öngörülebilen risklere, istenmeyen yan etkilere, kontrendikasyonlara ve uyarılara ilişkin bilgiler dâhil, fayda-risk analizinin ve risk yönetiminin özetini. 2.6. Bir insan kanı veya plazma türevi dâhil bir tıbbi madde ihtiva eden cihazlar ya da insan veya hayvan kaynaklı cansız dokular veya hücreler ya da bunların türevleri kullanılarak imal edilen cihazlar söz konusu olduğunda; cihazın klinik faydası ve/veya güvenliliği ile ilgili olarak bu tür bileşenleri ihtiva etmesinin artı değerine yönelik kanıtlar ile birlikte, tıbbi madde ya da dokular, hücreler veya bunların türevleri hakkında ayrıntılı bilgileri ve ilgili genel güvenlilik ve performans gerekliliklerine uygunluk ve maddeyle ya da dokular, hücreler veya bunların türevleriyle ilgili spesifik risk yönetimini. 2.7. Tamamen veya kısmen uygulanan standartlar ve ortak spesifikasyonlar dâhil, Ek I’de belirtilen ilgili genel güvenlilik ve performans gerekliliklerinin yerine getirilmesini ayrıntılayan bir liste ile birlikte bu standartların ve ortak spesifikasyonların yerine getirilmemesi veya yalnızca kısmen yerine getirilmiş olması ya da bu standartların ve ortak spesifikasyonların eksik olması durumunda ilgili genel güvenlilik ve performans gerekliliklerinin yerine getirilmesine yönelik çözümler ile ilgili bir açıklamayı. 2.8. Klinik araştırma esnasında kullanılan klinik prosedürlerin ve diyagnostik testlerin ayrıntılı bir açıklamasını ve özellikle normal klinik uygulamadan sapmalara ilişkin bilgileri. 3. Klinik Araştırma Planı Klinik araştırma planı (CIP); klinik araştırmaya yönelik gerekçeyi, amaçları, tasarım metodolojisini, izlemeyi, yürütmeyi, kayıt tutmayı ve analiz yöntemini düzenler. Bu plan, özellikle bu Ekte belirlenen bilgileri içerir. Bu bilgelerin bir bölümü ayrı bir dokümanda sunulursa, klinik araştırma planında bu dokümana atıfta bulunulur. 3.1. Genel 3.1.1. 70 inci maddenin birinci fıkrasında atıfta bulunulduğu şekilde, klinik araştırmanın tek kimlik numarası. 3.1.2. Sponsorun tanımlanması: sponsorun adı, adresi ve iletişim bilgileri ve mevcutsa sponsorun 62 nci maddenin ikinci fıkrası uyarınca yurt içinde yerleşik yasal temsilcisinin adı, adresi ve iletişim bilgileri. 3.1.3. Her araştırma merkezindeki sorumlu araştırmacı, araştırmanın koordinatör araştırmacısı, her araştırma merkezinin adresi ve her merkezdeki sorumlu araştırmacıya ilişkin acil iletişim detayları hakkında bilgiler. Araştırmacı tiplerinin görevleri, sorumlulukları ve nitelikleri, klinik araştırma planında belirtilir. 3.1.4. Klinik araştırmanın nasıl finanse edildiği ve sponsor ile araştırma merkezi arasındaki anlaşma ile ilgili kısa bir açıklama. 3.1.5. Klinik araştırmanın, Türkçe veya Kurum tarafından belirlenen AB resmî dillerinden birinde genel bir özeti. 3.2. Kullanım amacı, imalatçısı, izlenebilirliği, hedef popülasyonu, insan vücuduyla temas eden malzemeler, kullanımına dâhil edilen tıbbi veya cerrahi prosedürler ve kullanımı için gerekli eğitim ve deneyim, geçmiş literatür taraması, uygulamanın ilgili alanında klinik bakımdaki mevcut en son teknolojik gelişmeler ve yeni cihazın önerilen faydaları dâhil cihazın tanımlaması ve açıklaması. 3.3. İlgili beklenen klinik çıktıların klinik araştırma planında gerekçelendirilmesiyle birlikte, incelenecek cihazın riskleri ve klinik faydaları. 3.4. Klinik araştırmanın, klinik uygulamadaki en son gelişmelere dayandığına dair açıklama. 3.5. Klinik araştırmanın amaçları ve hipotezleri 3.6. Bilimsel sağlamlığının ve geçerliliğinin kanıtlarıyla birlikte klinik araştırmanın tasarımı. 3.6.1. Klinik değerlendirme planında belirtilen araştırma tipini seçmeye, sonlanım noktalarına ve değişkenlerine yönelik gerekçeler ile birlikte araştırma tipi gibi genel bilgiler. 3.6.2. Araştırma amaçlı cihaza, karşılaştırma cihazlarına ve klinik araştırmada kullanılacak diğer cihazlara veya ilaçlara ilişkin bilgiler. 3.6.3. Gönüllülere, seçim kriterlerine, araştırma popülasyonunun büyüklüğüne, hedef popülasyonla ilgili olarak araştırma popülasyonunun temsil edilebilirliğine ilişkin bilgiler ve mevcutsa çocuklar, gebe kadınlar, bağışıklık yetersizliği olan ya da ileri yaşta gönüllüler gibi araştırmaya dâhil edilen etkilenebilir gönüllülere ilişkin bilgiler. 3.6.4. Yanlılığı azaltmak için alınacak önlemlere (örn. randomizasyon) ilişkin ayrıntılar ve olası karışıklığa neden olan faktörlerin yönetimi. 3.6.5. Klinik araştırmayla ilgili klinik prosedürlerin ve diyagnostik yöntemlerin açıklanması ve özellikle normal klinik uygulamalardan sapmaların vurgulanması. 3.6.6. İzleme planı. 3.7. Mevcutsa örneklem büyüklüğüne yönelik bir güç hesaplaması da dâhil, gerekçeleriyle birlikte istatistiksel değerlendirmeler 3.8. Veri yönetimi. 3.9. Klinik araştırma planına yönelik değişikliklere ilişkin bilgiler. 3.10. Klinik araştırma planından sapmaların araştırma merkezinde takibi ile yönetimine ve klinik araştırma planına uyumsuzluğun açıkça yasaklanmasına ilişkin politika. 3.11. Cihaza, özellikle de cihaza erişim kontrolüne, klinik araştırmada kullanılan cihazla ilgili takibe ve kullanılmamış, süresi geçmiş veya arızalı cihazların iadesine ilişkin sorumluluk. 3.12. Uygulanabilir düzenleyici gerekliliklerin yanı sıra, insanların dâhil olduğu tıbbi araştırmaya yönelik kabul görmüş etik ilkelere ve cihazlarla ilgili klinik araştırmalar alanındaki “iyi klinik uygulamaları” ilkelerine uygunluk ile ilgili beyanname. 3.13. Bilgilendirilmiş gönüllü oluru sürecine ilişkin açıklama. 3.14. Advers olaylar ve ciddi advers olaylar, cihaz kusurları, prosedürler ve raporlamaya yönelik zaman çizelgeleri ile ilgili tanımlar dâhil, güvenlik raporlaması. 3.15. Bir araştırmanın tamamlanmasını, geçici olarak durdurulmasını veya erken sonlandırılmasını müteakip gönüllülerin takibi ile olurlarını geri çekmiş olan gönüllülerin takibine yönelik kriterler ve prosedürler ve takibi kaybedilen gönüllülere yönelik prosedürler. Bu tür prosedürler, implante edilebilir cihazlar için en azından izlenebilirliği kapsar. 3.16. Gönüllünün klinik araştırmaya katılımı nedeniyle ilave bakımın gerekli olması ve bu bakımın söz konusu tıbbi durum için normalde beklenenden farklı olması durumunda, klinik araştırmaya katılımlarının sonlanmasından sonra gönüllülerin bakımının üstlenilmesine yönelik düzenlemeler ile ilgili bir açıklama. 3.17. Klinik araştırma raporunun düzenlenmesi ve yasal gereklilikler ile bu Ekin I. Bölümünün 1 numaralı maddesinde atıfta bulunulan etik ilkeler uyarınca sonuçların yayımlanmasıyla ilgili politika. 3.18. Araştırmanın kapsamına giren özelliklere spesifik bir vurgu ile cihazın teknik ve işlevsel özelliklerinin listesi. 3.19. Kaynakça. 4. Diğer bilgiler 4.1. Araştırma amaçlı cihazın, klinik araştırma kapsamındaki hususlar haricinde genel güvenlilik ve performans gerekliliklerine uyduğuna ve bu hususlarla ilgili olarak bütün tedbirlerin gönüllünün sağlık ve güvenliğini korumak üzere alınmış olduğuna dair, söz konusu cihazın imalatından sorumlu gerçek veya tüzel kişi tarafından imzalı bir beyanname. 4.2. Uygulanabildiği hallerde; ilgili etik kurulun/kurulların görüşünün veya görüşlerinin sureti. Etik kurul/kurulların görüşünün veya görüşlerinin başvuru esnasında talep edilmemesi durumunda, görüş veya görüşlerin bir sureti, mümkün olan en kısa sürede sunulur. 4.3. 69 uncu madde ve ilgili mevzuat uyarınca, yaralanma durumunda gönüllülerin sigorta kapsamının veya teminatının kanıtını. 4.4. Hasta bilgilendirme föyü ve bilgilendirilmiş gönüllü oluru dokümanı dâhil, bilgilendirilmiş gönüllü oluru almak için kullanılacak dokümanları. 4.5. Kişisel verilerin korunmasına ve gizliliğine ilişkin uygulanabilir kurallara uyum için yapılan düzenlemeler ile ilgili açıklamayı, özellikle: - İşlenen bilgi ve kişisel verilere yetkisiz erişimi, bunların ifşasını, yayılmasını, değiştirilmesini veya kaybını önlemek için uygulanacak organizasyonel ve teknik düzenlemeleri, - Kayıtların ve gönüllülerin kişisel verilerinin gizliliğini sağlamak için uygulanacak önlemlerin açıklamasını ve - Bir veri güvenliği ihlali durumunda, olası advers etkileri azaltmak amacıyla uygulanacak önlemlerin açıklamasını. 4.6.Mevcut teknik dokümantasyonla ilgili, ayrıntılı risk analizi/yönetimi dokümantasyonu veya spesifik test raporları gibi tüm ayrıntılar, talebi üzerine başvuruyu gözden geçiren yetkili otoriteye sunulur.
III. BÖLÜM SPONSORUN DİĞER YÜKÜMLÜLÜKLERİ 1. Sponsor, bu Ekin II. Bölümünde atıfta bulunulan dokümantasyona yönelik kanıtlar sağlamak üzere gerekli her dokümantasyonu yetkili otorite için hazır bulundurmayı taahhüt eder. Sponsor, araştırma amaçlı cihazın imalatından sorumlu gerçek veya tüzel kişi değilse, bu yükümlülük, sponsor adına söz konusu gerçek veya tüzel kişi tarafından yerine getirilebilir. 2. Sponsor, 79 uncu maddenin ikinci fıkrasında atıfta bulunulduğu üzere ciddi advers olayların veya diğer olayların araştırmacı veya araştırmacılar tarafından sponsora zamanında raporlanmasını sağlamak için yürürlükte olan bir sözleşmeye sahip olur. 3. Bu Ekte bahsedilen dokümantasyon, söz konusu cihazın klinik araştırmasının sona ermesinden itibaren en az 10 yıllık ya da cihazın sonradan piyasaya arz edilmesi durumunda, en son cihaz piyasaya arz edildikten sonra en az 10 yıllık bir süre için muhafaza edilir. İmplante edilebilir cihazlar söz konusu olduğunda, bu süre en az 15 yıl olur. Kurum, yurt içinde yerleşik olan bir sponsorun veya 62 nci maddenin ikinci fıkrasında atıfta bulunulduğu üzere yurt içinde yerleşik olan yasal temsilcisinin, bu sürenin sona ermesinden önce iflas etmesi ya da faaliyetini sonlandırması durumunda, birinci alt paragrafta atıfta bulunulan süre boyunca bu dokümantasyonun yetkili otoritelere sunulmak üzere muhafaza edilmesini talep eder. 4. Sponsor; araştırmanın, klinik araştırma planı, iyi klinik uygulamaları ilkeleri ve bu Yönetmelik uyarınca yürütülmesini sağlamak için araştırma merkezinden bağımsız olan bir monitör (izleyici) atar. 5. Sponsor, araştırmaya katılan gönüllüler ile ilgili takip işlemlerini tamamlar. 6. Sponsor, araştırmanın iyi klinik uygulamaları doğrultusunda yürütülmekte olduğuna dair, örneğin iç ve dış denetim aracılığıyla, kanıtlar sağlar. 7. Sponsor, asgari olarak aşağıdakileri içeren bir klinik araştırma raporu hazırlar: - Araştırmanın başlığını, araştırma amaçlı cihazı, tek kimlik numarasını, klinik araştırma planı numarasını belirten kapak/giriş sayfası veya sayfalarını ve her araştırma merkezindeki koordinatör araştırmacı ve sorumlu araştırmacıların imzalarıyla birlikte ayrıntıları, - Raporun yazarı ile ilgili ayrıntıları ve tarihi, - Başlığı, araştırmanın amacını, araştırmanın açıklamasını, araştırmanın tasarımını ve kullanılan yöntemleri ve araştırmanın bulguları ile sonuçlarını içeren, araştırmanın bir özetini. Araştırmanın tamamlanma tarihi ve özellikle araştırmaların erken sonlandırılması, geçici durdurulması veya askıya alınması ile ilgili ayrıntıları, - Araştırma amaçlı cihazın açıklamasını, özellikle net bir şekilde tanımlanmış kullanım amacını, - Amaçlarını, tasarımını, etik yönlerini, izlenmesini ve kalite önlemlerini, seçim kriterlerini, hedef hasta popülasyonlarını, örneklem büyüklüğünü, tedavi planlarını, takip süresini, beraberinde alınan tedavileri ve hipotezi, örneklem büyüklüğü hesabını ve analiz yöntemleri dâhil istatistiksel planını ve gerekçeleri kapsayan, klinik araştırma planının bir özetini, - Klinik araştırma planına uygunluğun yanı sıra, gerekçesi ve sebebiyle birlikte gönüllü demografilerini, seçilmiş sonlanım noktaları ile ilgili sonuçların analizini, alt grup analizinin ayrıntılarını kapsayan ve eksik verilerin ve klinik araştırmadan erken çekilen hastaların takibini ya da takibi kaybedilenleri kapsayan klinik araştırma sonuçlarını. - Ciddi advers olayların, advers cihaz etkilerinin, cihaz kusurlarının ve ilgili düzeltici faaliyetlerin özetini, - Güvenlilik ve performans sonuçlarını, risklerin ve klinik faydaların değerlendirmesini, en son klinik gelişmeler ışığında klinik ilgililiğin tartışmasını, spesifik hasta popülasyonlarına yönelik spesifik tedbirleri, araştırma amaçlı cihaza yönelik tavsiyeleri, araştırmanın sınırlamalarını kapsayan tartışma ve genel değerlendirmeleri.
_______________________________________________
Ek XVI 1 İNCİ MADDENİN İKİNCİ FIKRASINDA ATIFTA BULUNULAN TIBBİ AMAÇLI OLMAYAN ÜRÜN GRUPLARININ LİSTESİ 1. Kontak lensler ya da göz içine veya üzerine uygulanması amaçlanan diğer gereçler. 2. Dövme ürünleri ve pirsingler (piercing) hariç olmak üzere, anatomiyi değiştirmek ya da vücut parçalarının fiksasyonu amacıyla, cerrahi invaziv yollar aracılığıyla insan vücuduna tamamen veya kısmen uygulanması amaçlanan ürünler. 3. Dövmeye yönelik olanlar hariç olmak üzere, subkütan, submukoz veya intradermal enjeksiyon ya da başka bir uygulama yoluyla fasiyal veya diğer dermal dolgu ya da mukoz membran dolgusu olarak kullanılması amaçlanan maddeler, maddelerin kombinasyonları ya da gereçler. 4. Liposakşın, lipoliz veya lipoplastiye yönelik ekipman gibi, yağ dokusunu azaltmak, uzaklaştırmak veya parçalamak için kullanılması amaçlanan ekipman. 5. Cilt yenilemeye, dövme silme veya tüy almaya ya da diğer cilt uygulamalarına yönelik lazerler ve yoğun atımlı ışık (IPL) ekipmanı gibi, monokromatik ve geniş spektrumda, eş fazlı ve eş fazlı olmayan kaynaklar dâhil, insan vücudu üzerinde kullanılması amaçlanan yüksek yoğunluklu elektromanyetik radyasyon (örneğin, kızıl-ötesi/infrared, görünür ışık ve ultraviyole) yayan ekipman. 6. Beyindeki sinirsel aktiviteyi değiştirmek için kafatasına penetre olan elektrik akımları ya da manyetik veya elektromanyetik alanlar uygulayan beyin stimülasyonu amaçlı ekipman.
|
|